Inhibitor of apoptosis
Inhibitors of apoptosis are a group of proteins that mainly act on the intrinsic pathway that block programmed cell death, which can frequently lead to cancer or other effects for the cell if mutated or improperly regulated. Many of these inhibitors act to block caspases, a family of cysteine proteases that play an integral role in apoptosis. Some of these inhibitors include the Bcl-2 family, viral inhibitor crmA, and IAP's.
Apoptosis, or programmed cell death, is a highly regulated process used by many multicellular organisms. Like any regulated process, apoptosis is subject to either activation or inhibition by a variety of chemical factors. Apoptosis can be triggered through two main pathways; extrinsic and intrinsic. The extrinsic pathway mostly involves extracellular signals triggering intracellular apoptosis mechanisms by binding to receptors in the cell membrane and sending signals from the outside of the cell. Intrinsic pathways involved internal cell signaling primarily through the mitochondria.[1]
Bcl-2 family
The Bcl-2 family of proteins can either inhibit or promote apoptosis and members are characterized by the Bcl-2 homologous domains BH1, BH2, BH3, and BH4. The combinations of the domains in the proteins determine its role in the apoptosis process. Members of the family that inhibit apoptosis include Bcl-2 itself, Bcl-XL, and Bcl-w, which possess all four of the domains.[2] Bcl-2 is the most well known of the anti-apoptotic members, and is classified as an oncogene. Studies have shown that the Bcl-2 oncogene may inhibit apoptosis in two ways; either by directly controlling the activation of caspases, or by disrupting the channels that allow proapoptotic factors from leaving the mitochondria.
Activity in the cell
Regarding the activation of caspases, there exists a gene called ced-9 in C. elegans that protects against cell death that is a part of the Bcl-2 family. ced-9 encodes a protein that is structurally similar to Bcl-2 that binds to another protein ced-4, a homolog of APAF-1 in humans, and prevents it from activating caspase ced-3, which is necessary for killing of the cell.[3] In humans APAF-1 actually doesn't interact with Bcl-2; rather it is activated by cytochrome c, the release of which from the mitochondria is regulated by Bcl-2. BAX and BAK are multidomain proapoptoic members of the Bcl-2 family that lie in the cytosol and the mitochondria, respectively. After several stimuli leading to cell death are activated, BAX also moves to the mitochondria where it carries out its functions there. Bcl-2 and Bcl-xl have been found to sequester BH3 domain molecules in the mitochondria, which prevents the activation of BAX and BAK.[4]
crmA
| Cytokine response modifier A | |||||||
|---|---|---|---|---|---|---|---|
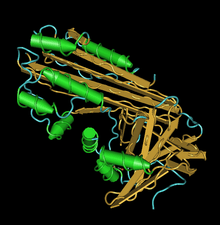 crmA structure with visible domain | |||||||
| Identifiers | |||||||
| Organism | |||||||
| Symbol | CrmA | ||||||
| Alt. symbols | Serine proteinase inhibitor 2 | ||||||
| Entrez | 1486086 | ||||||
| RefSeq (Prot) | NP_619988.1 | ||||||
| UniProt | P07385 | ||||||
| Other data | |||||||
| Chromosome | genomic: 0.19 - 0.19 Mb | ||||||
| |||||||
crmA, or cytokine response modifier A, is a cowpox serpin that inhibits caspases 1, 6 and 8, forming complexes with these caspases that renders them unable to perform their apoptotic functions. Cowpox is an orthopox virus that increases their chances of survival and infection by inhibition of specific caspases and preventing inflammatory responses and apoptosis.[5]
- By inhibiting caspase 1, also known as interleukin 1β converting enzyme (ICE), crmA prevents cytokines interleukin 1β from being formed, preventing an inflammatory response. Although free floating crmA is relatively unstable, it becomes incredibly stable after binding to caspase 1, forming an irreversible stable complex.[6]
- Caspase 8 initiates apoptosis by activating "executioner" caspases, numbered 3, 6, and 7. By inhibiting caspase 8, crmA prevents the other caspases from ever being activated. Inhibition of caspase 8 also prevents cell death signals by ligation of a TNF super family member known as death receptors, that signal for apoptosis through caspase 8.[6]
Serpins generally inhibit serine proteases by a suicide substrate inhibition mechanism, in which the serpin undergoes a drastic change in structure to form an acyl enzyme intermediate. A reactive center loop of the protease is bound to the central beta loop of the serpin, trapping the protease in a state where it is no longer able to perform its catalytic functions. Studies have shown crmA uses a method analogous to serpin inhibition of serine proteases to inhibit cysteine protease caspases.[5]
IAPs
The Inhibitors of apoptosis proteins (IAP) are a family of functionally and structurally related proteins that serve as endogenous inhibitors of programmed cell death (apoptosis). A common feature of all IAPs is the presence of a BIR (Baculovirus IAP Repeat, a ~70 amino acid domain) in one to three copies. The human IAP family consists of 8 members, and IAP homologs have been identified in numerous organisms.
The first members of the IAPs identified were from the baculovirus IAPs, Cp-IAP and Op-IAP, which bind to and inhibit caspases as a mechanism that contributes to its efficient infection and replication cycle in the host. Later, five more human IAPs were discovered which included XIAP, cIAP1 , C-IAP2, NAIP, Livin and Survivin.
The best characterized IAP is XIAP, which binds caspase-9, caspase-3 and caspase-7, thereby inhibiting their activation and preventing apoptosis. Also cIAP1 and cIAP2 have been shown to bind caspases, although how the IAPs inhibit apoptosis mechanistically at the molecular level is not completely understood.
Activity of XIAP is blocked by binding to DIABLO (Smac) and HTRA2 (Omi) proteins released from mitochondria after proapoptotic stimuli.[7]
Since the mid 2000s, significant progress has been made into the development of small molecule mimics of the endogenous IAP ligand Smac. One recent example published in 2013 describes the synthesis and testing of peptidomimetics whose structure is based on the AVPI tetrapeptide IAP binding motif present in the N-terminus of mature Smac. These peptidomimetic compounds were specifically noted for their exceptionally high level of binding to Livin, one of the important IAP family members yet to receive much attention from a drug discovery perspective.[8]
LCL161 is a drug that promotes cancer cell death by antagonizing IAPs. Clinical trials are ongoing, but have received mixed response. A Phase I clinical trial determined that LCL161 is well tolerated in the treatment of patients with advanced solid tumors, though they experienced vomiting, nausea, fatigue, and loss of appetite.[9] Another study found that LCL161 reduced survival and promoted endotoxic shock when used in MYC-driven lymphoma.[10] Other clinical trials are still enrolling to determine the drugs efficacy.[11]
References
- Schwerk C, Schulze-Osthoff K (July 2005). "Regulation of apoptosis by alternative pre-mRNA splicing". Molecular Cell. 19 (1): 1–13. doi:10.1016/j.molcel.2005.05.026. PMID 15989960.
- Mayer B, Oberbauer R (2003). "Mitochondrial regulation of apoptosis". News in Physiological Sciences. 18: 89–94. doi:10.1152/nips.01433.2002. PMID 12750442.
- Conradt B, Horvitz HR (1998). "The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9". Cell. 93 (4): 519–29. doi:10.1016/S0092-8674(00)81182-4. PMID 9604928.
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001). "BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis". Molecular Cell. 8 (3): 705–11. doi:10.1016/S1097-2765(01)00320-3. PMID 11583631.
- Dobó J, Swanson R, Salvesen GS, Olson ST, Gettins PG (December 2006). "Cytokine response modifier a inhibition of initiator caspases results in covalent complex formation and dissociation of the caspase tetramer". The Journal of Biological Chemistry. 281 (50): 38781–90. doi:10.1074/jbc.M605151200. PMID 17052983.
- Callus BA, Vaux DL (January 2007). "Caspase inhibitors: viral, cellular and chemical". Cell Death and Differentiation. 14 (1): 73–8. doi:10.1038/sj.cdd.4402034. PMID 16946729.
- Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC (1998). "A single BIR domain of XIAP sufficient for inhibiting caspases". The Journal of Biological Chemistry. 273 (14): 7787–90. doi:10.1074/jbc.273.14.7787. PMID 9525868.
- Vamos M, Welsh K, Finlay D, Lee PS, Mace PD, Snipas SJ, Gonzalez ML, Ganji SR, Ardecky RJ, Riedl SJ, Salvesen GS, Vuori K, Reed JC, Cosford ND (April 2013). "Expedient synthesis of highly potent antagonists of inhibitor of apoptosis proteins (IAPs) with unique selectivity for ML-IAP". ACS Chemical Biology. 8 (4): 725–32. doi:10.1021/cb3005512. PMC 3953502. PMID 23323685.
- Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, Cohen RB (October 2014). "Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors". Journal of Clinical Oncology. 32 (28): 3103–10. doi:10.1200/JCO.2013.52.3993. PMID 25113756.
- West AC, Martin BP, Andrews DA, Hogg SJ, Benerjee A, Grigoriadis G, Johnstone RW, Shortt J (2016). "The SMAC mimetic, LCL-161, reduces survival in aggressive MYC-driven lymphoma while promoting susceptibility to endotoxic shock". Oncogenesis. 5: e216. doi:10.1038/oncsis.2016.26. PMC 4848837. PMID 27043662.
- "Clinical trials using Smac Mimetic LCL161". National Cancer Institute. Retrieved April 20, 2018.
External links
- The MEROPS online database for peptidases and their inhibitors: I32.002
- Inhibitor+of+Apoptosis+Proteins at the US National Library of Medicine Medical Subject Headings (MeSH)