Clonal hematopoiesis
Clonal hematopoiesis of indeterminate potential, or CHIP, is a common aging-related phenomenon in which hematopoietic stem cells (HSCs) or other early blood cell progenitors contribute to the formation of a genetically distinct subpopulation of blood cells.[1][2][3] As the name suggests, this subpopulation in the blood is characterized by a shared unique mutation in the cells' DNA; it is thought that this subpopulation is "clonally" derived from a single founding cell and is therefore made of genetic "clones" of the founder.[4][5][6][7] The establishment of a clonal population may occur when a stem or progenitor cell acquires one or more somatic mutations that give it a competitive advantage in hematopoiesis over the stem/progenitor cells without these mutations.[1][3] Alternatively, clonal hematopoiesis may arise without a driving mutation, through mechanisms such as neutral drift in the stem cell population.[8] Clonal hematopoiesis may occur in people who are completely healthy but has also been found in people with hematologic diseases.[1][9][10] The clonal population may vary in size depending on the person, where it can be less than 2% of the blood or, at the other end, can sometimes grow close to 100%.[4][9] The incidence of clonal hematopoiesis has been found to rise dramatically with age. Recent studies have demonstrated that less than 1% of the population under age 40 but approximately 10-20% of the population over age 70 has observable clonal hematopoiesis.[4][5][6] Having clonal hematopoiesis has been linked to a more than 10-fold increased risk of developing a blood cancer, though the overall likelihood is still low.[4][5] Clonal hematopoiesis does not typically give rise to noticeable symptoms, but does lead to increased risk of cardiovascular disease.[1][5][11]
History
The first major evidence for the existence of prevalent clonal hematopoiesis in healthy people was put forth in the 1990s. Using the HUMARA assay, scientists found that there was nonrandom X-inactivation of the X chromosome in the blood of some healthy women.[12][13] This means that a greater than expected proportion of the blood had the silencing of one specific X chromosome in the chromosome pair. Just as the observation of the same DNA mutation in a subset of cells suggests a single founding source, this X-inactivation skew suggests that a greater than expected number of cells are being generated from the same precursor. Importantly, these findings described an increase in this nonrandom skewing with increasing age, hinting that unobserved mutations acquired with age could be driving a clonal expansion. In a similar vein, other studies using the HUMARA technology had found that hematologic malignancies are clonal diseases even when there is no apparent chromosomal abnormality,[14][15] and that there are pre-leukemic clonal populations which precede acute myeloid leukemia (AML).[16] As the HUMARA assay is based on the epigenetic state of cells, the underlying genetic determinants of the clonal expansion remained to be uncovered.
This set of evidence led to the suggestion in 2005 that driving mutations in leukemia are acquired in a step-wise manner.[17] This model has received support from studies showing subpopulations of blood cells harboring initiating but not late somatic mutations in patients with chronic lymphocytic leukemia (CLL),[18][19] hairy cell leukemia (HCL),[20] and AML.[21][22][23]
The combination of these two ideas, that clonal hematopoiesis might be common in the elderly population and that AML evolves from pre-leukemic populations, led to the hypothesis that malignancy-associated mutations could also contribute to asymptomatic clonal hematopoiesis in healthy individuals.[1] This view gained mechanistic support in 2012 when it was found a number of the women who showed evidence for clonal hematopoiesis through X-inactivation skew also had mutations in the hematologic-malignancy-associated gene TET2.[24]
Recently, several independent studies have confirmed the presence of malignancy-associated mutations in the blood of individuals who have no clinical signs of hematologic malignancy.[4][5][6] In combination, these studies have demonstrated the widespread incidence of clonal hematopoiesis in the healthy adult population and have stimulated further efforts to broaden our understanding of clonal hematopoiesis in health and disease.
Population genetics
The advent of next-generation DNA sequencing has allowed for the targeted identification of somatic mutations involved in clonal hematopoiesis at the population level. The studies undertaken as of 2017 are largely consistent in their main findings. One common finding has been that observable clonal hematopoiesis is virtually absent from the under-40 population, with a sharp uptick in frequency past 60 years of age.[4][5][6] Indeed, the evidence from these studies suggests that between 10% and 20% of the population over age 70 have clonal hematopoiesis. In the U.S. alone, this means that, at the low end, some 2,975,000 seniors over 70 years of age are living with this condition.[25]
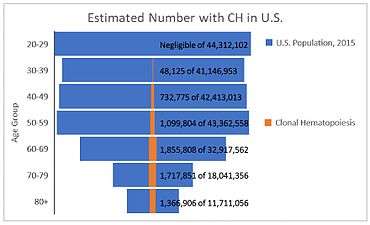
The other main common finding is that there are many different mutations involved in clonal hematopoiesis. Many of these fall into the categories of epigenetic regulators (DNMT3a, TET2, and ASXL1), signaling proteins (JAK2), spliceosome components (SF3B1 and SRSF2), or members of the DNA damage response (TP53 and PPM1D).[4][5][6] Many people identified as having clonal hematopoiesis have a mutation in a single gene, though a significant number have mutations in two or more genes.[4][5][6] The number and variety of observed mutations suggests that these mutations may contribute to clonal hematopoiesis by several distinct mechanisms, discussed in more detail below.
There is also limited evidence suggesting clonal hematopoiesis may be ubiquitous in healthy adults, albeit at extremely low levels (less than 0.1% of peripheral blood cells). A study employing the ultra-sensitive digital droplet PCR method found that 95% of studied individuals (19 out of 20) between the ages of 50 and 70 had at least low-level clonal hematopoiesis.[26] This finding does not necessarily conflict with earlier reports that clonal hematopoisis is not ubiquitous in this age bracket, as these previous studies' experimental designs compels the use of a higher threshold to identify legitimate clonal hematopoiesis.[4][5][6]
As of 2017, there is little known about which, if any, genetic and epidemiological factors may influence the acquisition of mutations in clonal hematopoiesis.[1] One study found that there was a strong association between smoking and clonal hematopoiesis.[4] However, as this result was obtained in a diagnosis-specific all-Swedish cohort, it is unclear how generalizable this result will ultimately be.
Biology
Clonal hematopoiesis is thought to originate with the hematopoietic stem cells that make blood. An adult human has approximately 10,000 to 20,000 HSCs.[27] The fact that these cells are maintained for life and each HSC may acquire about one mutation in a protein-coding exon each decade[28] means that an elderly individual will have a certain amount of genetic mosaicism, or a variety of cells with different unique mutations, within their HSC population. However, this does not lead to clonal hematopoiesis in all cases. It is only when the genetic mutation confers a selective advantage on its host or there is another favorable stem cell dynamic that there is a clonal expansion.
Candidate driver mutations
There are several general mechanisms by which a mutation could provide such an advantage and it is likely that the mutations found in clonal hematopoiesis act through different pathways. First, a mutation could provide a growth advantage, causing HSCs to divide more rapidly and contribute a larger proportion of the mature blood cells. This may be the case for mutations in genes related to signaling, such as that which causes the activating V617F substitution in the JAK2 signaling protein. Mutations in the DNA damage response genes would appear more likely to act via a second mechanism: allowing for HSC survival and proliferation under normally lethal cytotoxic stress.[1]
Other mechanisms are more likely to be associated with the disruption of epigenetic regulators, which comprises 80% of observed mutations in clonal hematopoiesis. A third potential mechanism of action is that the mutation makes the HSC-derived progenitor cells less able to differentiate into mature blood cells. This would allow these cells to continue to divide even after they would have normally stopped, since progenitor cells may divide whereas normal mature blood cells cannot. A fourth possibility is that the mutation makes the progenitor cells and cells derived from them more like stem cells in their ability to keep dividing. The previous two possibilities are very similar in terms of physiologic outcome and mainly differ on what is happening at the DNA level: whether differentiation genes are suppressed or a stem cell program is upregulated. A final possibility is that a gradient of epigenetic states is created in the HSC and progenitor cells and the cells with the most favorable epigenetics are able to grow out faster than unmutated cells.[1]
Non-candidate-driver mechanisms
An expansion of blood cells from a single source does not necessarily require a mutation to act as the driving force. A large proportion of the population who exhibit clonal hematopoiesis have no identifiable mutations in known candidate driver genes.[4][8] One possible explanation is that among a naturally-occurring specturm of inheritable epigenetic states, there are those which augment the self-renewal or proliferation of a stem cell and its progeny.[8] Another explanation is that a process of neutral drift causes the predominance of a clonal stem cell population over time. In this scenario, all stem cells have an equal proliferative potential but some of them die out in a stochastic manner leading some of the remaining cells to proliferate to replace them.[8][29] This can be equated to a game of chance where all players start with the same odds of winning. As the game is played, winners and losers will arise despite the equal starting positions.[29]
Implications for human health
Clonal hematopoiesis by itself is not considered to be a hematologic cancer; nevertheless, evidence is mounting that this condition may adversely affect human health. It has been proposed to label the group of individuals who have clonal hematopoiesis defined by a mutation in a malignancy-associated gene but without evidence of disease (such as cytopenia, dysplasia or immature “blast” cells in the bone marrow) as having Clonal Hematopoiesis of Indeterminate Potential (CHIP).[1][3][30] A clonal involvement (sometimes referred to simply as the size of a “clone”) of 2% of the blood has been tentatively proposed as a cutoff, though there is discussion that a lower floor that is more inclusive could also be appropriate.[1][3][26][31] This cutoff may ultimately depend on whether clones must reach a certain size before influencing health. The level at which a clone begins to have a potential clinical impact is an open question, though there is already data to suggest larger clones have a larger effect on health.[5]
The presence of clonal hematopoiesis/CHIP has been shown to increase blood cancer risk and is correlated with an increased risk of mortality overall.[4][5][8] This is true both of clonal hematopoiesis with known candidate drivers as well as in cases without such drivers.[8]
Blood cancer risk
One area of health that CHIP has been definitively shown to influence is the risk of progression to blood cancer. In a given year, a tiny fraction of the general population will develop a hematologic cancer such as myelodysplastic syndrome (MDS) or AML; it is estimated that just 3 to 4 people per 100,000 will get MDS in a given year,[32] and 4 people per 100,000 will develop AML.[33] With CHIP, the risk of acquiring a hematologic malignancy like MDS or AML is increased more than 10-fold.[4][5] Despite this increased risk, people with CHIP are still at low overall risk for developing a blood cancer, with only about 0.5-1.0% transformation per year.[1]
Cardiovascular risk
A second area of health that may be affected by CHIP is the risk for heart attack and stroke. A strong association between CHIP and heart attack/ischemic stroke has been identified in one human genetic dataset, where CHIP was a stronger predictor of heart attack/stroke than if a patient 1) was a smoker, 2) had hypertension, 3) had high cholesterol, or 4) was overweight. In this study, which shows correlation but not causation, people with CHIP were 2.3 times more likely to suffer a heart attack, or 4.4 times as likely if the variant allele frequency in their blood was greater than 0.10, than matched controls without CHIP.[5] It has also been found that there is an increased risk of cardiovascular mortality in patients who exhibit CHIP and receive self-derived stem cell transplantation.[10] The idea of CHIP having a causal role in human heart attacks/strokes has been given support by a 2017 study that showed impairment of the Tet2 CHIP gene in mice causally led to accelerated atherosclerosis,[34] and this finding in mice has been independently validated.[11] The possibility of somatic mutations in the blood contributing not only to cancer risk but also to heart attack and stroke has generated much discussion in top-level scientific publications[35][36] and a large multi-cohort study published in 2017 appears to confirm the causal link between CHIP and cardiovascular disease in humans.[11]
Comorbidities
In addition to its effects on those who would otherwise be considered healthy, CHIP may have implications in certain disease contexts. It has been shown that patients with CHIP who receive autologous stem cell transplantation (ASCT) as part of their treatment for lymphoma have worse outcomes than patients without CHIP. The poorer prognosis for these patients is due to both an increase in subsequent therapy-related myeloid neoplasms and increased risk for cardiovascular mortality.[10]
Treatment
There are currently no therapies for slowing or targeting CHIP mutations. Together with the fact that progression from CHIP to outright hematologic malignancy remains infrequent, medical experts have argued against preemptive screening for CHIP but suggest routine follow-up for incidental CHIP findings.[1][3]
Associated disorders
Clonal hematopoiesis is sometimes compared to the unrelated blood disorders of monoclonal gammopathy of undetermined significance (MGUS) and monoclonal B-cell lymphocytosis (MBL) to which it bears similarities in its apparent priming for more advanced hematologic disease combined with a lack of symptoms and overall low risk of progression.[1][3] The acquisition of additional mutations can cause CHIP to transform into the related blood disorders MDS and AML.[3][30]
See also
References
- Jan, Max; Ebert, Benjamin L.; Jaiswal, Siddhartha (1 January 2017). "Clonal hematopoiesis". Seminars in Hematology. 54 (1): 43–50. doi:10.1053/j.seminhematol.2016.10.002. ISSN 1532-8686. PMID 28088988.
- Sperling, Adam S.; Gibson, Christopher J.; Ebert, Benjamin L. (2017). "The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia". Nature Reviews Cancer. 17 (1): 5–19. doi:10.1038/nrc.2016.112. ISSN 1474-1768. PMC 5470392. PMID 27834397.
- Steensma, David P.; Bejar, Rafael; Jaiswal, Siddhartha; Lindsley, R. Coleman; Sekeres, Mikkael A.; Hasserjian, Robert P.; Ebert, Benjamin L. (2 July 2015). "Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes". Blood. 126 (1): 9–16. doi:10.1182/blood-2015-03-631747. ISSN 1528-0020. PMC 4624443. PMID 25931582.
- Genovese, Giulio; Kähler, Anna K.; Handsaker, Robert E.; Lindberg, Johan; Rose, Samuel A.; Bakhoum, Samuel F.; Chambert, Kimberly; Mick, Eran; Neale, Benjamin M.; Fromer, Menachem; Purcell, Shaun M.; Svantesson, Oscar; Landén, Mikael; Höglund, Martin; Lehmann, Sören; Gabriel, Stacey B.; Moran, Jennifer L.; Lander, Eric S.; Sullivan, Patrick F.; Sklar, Pamela; Grönberg, Henrik; Hultman, Christina M.; McCarroll, Steven A. (25 December 2014). "Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence". The New England Journal of Medicine. 371 (26): 2477–2487. doi:10.1056/NEJMoa1409405. ISSN 1533-4406. PMC 4290021. PMID 25426838.
- Jaiswal, Siddhartha; Fontanillas, Pierre; Flannick, Jason; Manning, Alisa; Grauman, Peter V.; Mar, Brenton G.; Lindsley, R. Coleman; Mermel, Craig H.; Burtt, Noel; Chavez, Alejandro; Higgins, John M.; Moltchanov, Vladislav; Kuo, Frank C.; Kluk, Michael J.; Henderson, Brian; Kinnunen, Leena; Koistinen, Heikki A.; Ladenvall, Claes; Getz, Gad; Correa, Adolfo; Banahan, Benjamin F.; Gabriel, Stacey; Kathiresan, Sekar; Stringham, Heather M.; McCarthy, Mark I.; Boehnke, Michael; Tuomilehto, Jaakko; Haiman, Christopher; Groop, Leif; Atzmon, Gil; Wilson, James G.; Neuberg, Donna; Altshuler, David; Ebert, Benjamin L. (25 December 2014). "Age-related clonal hematopoiesis associated with adverse outcomes". The New England Journal of Medicine. 371 (26): 2488–2498. doi:10.1056/NEJMoa1408617. ISSN 1533-4406. PMC 4306669. PMID 25426837.
- Xie, Mingchao; Lu, Charles; Wang, Jiayin; McLellan, Michael D.; Johnson, Kimberly J.; Wendl, Michael C.; McMichael, Joshua F.; Schmidt, Heather K.; Yellapantula, Venkata; Miller, Christopher A.; Ozenberger, Bradley A.; Welch, John S.; Link, Daniel C.; Walter, Matthew J.; Mardis, Elaine R.; Dipersio, John F.; Chen, Feng; Wilson, Richard K.; Ley, Timothy J.; Ding, Li (1 December 2014). "Age-related mutations associated with clonal hematopoietic expansion and malignancies". Nature Medicine. 20 (12): 1472–1478. doi:10.1038/nm.3733. ISSN 1546-170X. PMC 4313872. PMID 25326804.
- McKerrell, T; Park, N; Moreno, T; Grove, CS; Ponstingl, H; Stephens, J; Understanding Society Scientific, Group.; Crawley, C; Craig, J; Scott, MA; Hodkinson, C; Baxter, J; Rad, R; Forsyth, DR; Quail, MA; Zeggini, E; Ouwehand, W; Varela, I; Vassiliou, GS (3 March 2015). "Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis". Cell Reports. 10 (8): 1239–45. doi:10.1016/j.celrep.2015.02.005. PMC 4542313. PMID 25732814.
- Zink, Florian; Stacey, Simon N.; Norddahl, Gudmundur L.; Frigge, Michael L.; Magnusson, Olafur T.; Jonsdottir, Ingileif; Thorgeirsson, Thorgeir E.; Sigurdsson, Asgeir; Gudjonsson, Sigurjon A. (2017-01-01). "Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly". Blood. 130 (6): blood–2017–02–769869. doi:10.1182/blood-2017-02-769869. ISSN 0006-4971. PMC 5553576. PMID 28483762.
- da Silva-Coelho, Pedro; Kroeze, Leonie I.; Yoshida, Kenichi; Koorenhof-Scheele, Theresia N.; Knops, Ruth; van de Locht, Louis T.; de Graaf, Aniek O.; Massop, Marion; Sandmann, Sarah; Dugas, Martin; Stevens-Kroef, Marian J.; Cermak, Jaroslav; Shiraishi, Yuichi; Chiba, Kenichi; Tanaka, Hiroko; Miyano, Satoru; de Witte, Theo; Blijlevens, Nicole M. A.; Muus, Petra; Huls, Gerwin; van der Reijden, Bert A.; Ogawa, Seishi; Jansen, Joop H. (21 April 2017). "Clonal evolution in myelodysplastic syndromes". Nature Communications. 8: 15099. doi:10.1038/ncomms15099. ISSN 2041-1723. PMC 5530598. PMID 28429724.
- Gibson, Christopher J.; Lindsley, R. Coleman; Tchekmedyian, Vatche; Mar, Brenton G.; Shi, Jiantao; Jaiswal, Siddhartha; Bosworth, Alysia; Francisco, Liton; He, Jianbo; Bansal, Anita; Morgan, Elizabeth A.; Lacasce, Ann S.; Freedman, Arnold S.; Fisher, David C.; Jacobsen, Eric; Armand, Philippe; Alyea, Edwin P.; Koreth, John; Ho, Vincent; Soiffer, Robert J.; Antin, Joseph H.; Ritz, Jerome; Nikiforow, Sarah; Forman, Stephen J.; Michor, Franziska; Neuberg, Donna; Bhatia, Ravi; Bhatia, Smita; Ebert, Benjamin L. (9 January 2017). "Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma". Journal of Clinical Oncology. 35 (14): 1598–1605. doi:10.1200/JCO.2016.71.6712. ISSN 1527-7755. PMC 5455707. PMID 28068180.
- Jaiswal, Siddhartha; Natarajan, Pradeep; Silver, Alexander J.; Gibson, Christopher J.; Bick, Alexander G.; Shvartz, Eugenia; McConkey, Marie; Gupta, Namrata; Gabriel, Stacey (2017-06-21). "Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease". New England Journal of Medicine. 0 (2): 111–121. doi:10.1056/NEJMoa1701719. ISSN 0028-4793. PMC 6717509. PMID 28636844.
- Busque, L.; Mio, R.; Mattioli, J.; Brais, E.; Blais, N.; Lalonde, Y.; Maragh, M.; Gilliland, D. G. (1 July 1996). "Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age". Blood. 88 (1): 59–65. doi:10.1182/blood.V88.1.59.59. ISSN 0006-4971. PMID 8704202.
- Champion, K. M.; Gilbert, J. G.; Asimakopoulos, F. A.; Hinshelwood, S.; Green, A. R. (1 June 1997). "Clonal haemopoiesis in normal elderly women: implications for the myeloproliferative disorders and myelodysplastic syndromes". British Journal of Haematology. 97 (4): 920–926. doi:10.1046/j.1365-2141.1997.1933010.x. ISSN 0007-1048. PMID 9217198.
- Fialkow, P. J.; Singer, J. W.; Adamson, J. W.; Berkow, R. L.; Friedman, J. M.; Jacobson, R. J.; Moohr, J. W. (5 July 1979). "Acute nonlymphocytic leukemia: expression in cells restricted to granulocytic and monocytic differentiation". The New England Journal of Medicine. 301 (1): 1–5. doi:10.1056/NEJM197907053010101. ISSN 0028-4793. PMID 286882.
- Fialkow, P. J.; Singer, J. W.; Adamson, J. W.; Vaidya, K.; Dow, L. W.; Ochs, J.; Moohr, J. W. (1 June 1981). "Acute nonlymphocytic leukemia: heterogeneity of stem cell origin". Blood. 57 (6): 1068–1073. doi:10.1182/blood.V57.6.1068.bloodjournal5761068. ISSN 0006-4971. PMID 6939452.
- Fialkow, P. J.; Janssen, J. W.; Bartram, C. R. (1 April 1991). "Clonal remissions in acute nonlymphocytic leukemia: evidence for a multistep pathogenesis of the malignancy". Blood. 77 (7): 1415–1417. doi:10.1182/blood.V77.7.1415.1415. ISSN 0006-4971. PMID 2009365.
- Weissman, Irving (21 September 2005). "Stem cell research: paths to cancer therapies and regenerative medicine". JAMA. 294 (11): 1359–1366. doi:10.1001/jama.294.11.1359. ISSN 1538-3598. PMID 16174694.
- Kikushige, Yoshikane; Ishikawa, Fumihiko; Miyamoto, Toshihiro; Shima, Takahiro; Urata, Shingo; Yoshimoto, Goichi; Mori, Yasuo; Iino, Tadafumi; Yamauchi, Takuji; Eto, Tetsuya; Niiro, Hiroaki; Iwasaki, Hiromi; Takenaka, Katsuto; Akashi, Koichi (16 August 2011). "Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia". Cancer Cell. 20 (2): 246–259. doi:10.1016/j.ccr.2011.06.029. ISSN 1878-3686. PMID 21840488.
- Damm, Frederik; Mylonas, Elena; Cosson, Adrien; Yoshida, Kenichi; Della Valle, Véronique; Mouly, Enguerran; Diop, M'boyba; Scourzic, Laurianne; Shiraishi, Yuichi; Chiba, Kenichi; Tanaka, Hiroko; Miyano, Satoru; Kikushige, Yoshikane; Davi, Frederick; Lambert, Jérôme; Gautheret, Daniel; Merle-Béral, Hélène; Sutton, Laurent; Dessen, Philippe; Solary, Eric; Akashi, Koichi; Vainchenker, William; Mercher, Thomas; Droin, Nathalie; Ogawa, Seishi; Nguyen-Khac, Florence; Bernard, Olivier A. (1 September 2014). "Acquired initiating mutations in early hematopoietic cells of CLL patients". Cancer Discovery. 4 (9): 1088–1101. doi:10.1158/2159-8290.CD-14-0104. ISSN 2159-8290. PMID 24920063.
- Chung, Stephen S.; Kim, Eunhee; Park, Jae H.; Chung, Young Rock; Lito, Piro; Teruya-Feldstein, Julie; Hu, Wenhuo; Beguelin, Wendy; Monette, Sebastien; Duy, Cihangir; Rampal, Raajit; Telis, Leon; Patel, Minal; Kim, Min Kyung; Huberman, Kety; Bouvier, Nancy; Berger, Michael F.; Melnick, Ari M.; Rosen, Neal; Tallman, Martin S.; Park, Christopher Y.; Abdel-Wahab, Omar (28 May 2014). "Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia". Science Translational Medicine. 6 (238): 238ra71. doi:10.1126/scitranslmed.3008004. ISSN 1946-6242. PMC 4501573. PMID 24871132.
- Jan, Max; Snyder, Thomas M.; Corces-Zimmerman, M. Ryan; Vyas, Paresh; Weissman, Irving L.; Quake, Stephen R.; Majeti, Ravindra (29 August 2012). "Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia". Science Translational Medicine. 4 (149): 149ra118. doi:10.1126/scitranslmed.3004315. ISSN 1946-6242. PMC 4045621. PMID 22932223.
- Corces-Zimmerman, M. Ryan; Hong, Wan-Jen; Weissman, Irving L.; Medeiros, Bruno C.; Majeti, Ravindra (18 February 2014). "Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission". Proceedings of the National Academy of Sciences of the United States of America. 111 (7): 2548–2553. doi:10.1073/pnas.1324297111. ISSN 1091-6490. PMC 3932921. PMID 24550281.
- Shlush, Liran I.; Zandi, Sasan; Mitchell, Amanda; Chen, Weihsu Claire; Brandwein, Joseph M.; Gupta, Vikas; Kennedy, James A.; Schimmer, Aaron D.; Schuh, Andre C.; Yee, Karen W.; McLeod, Jessica L.; Doedens, Monica; Medeiros, Jessie J. F.; Marke, Rene; Kim, Hyeoung Joon; Lee, Kwon; McPherson, John D.; Hudson, Thomas J.; Brown, Andrew M. K.; Yousif, Fouad; Trinh, Quang M.; Stein, Lincoln D.; Minden, Mark D.; Wang, Jean C. Y.; Dick, John E. (20 February 2014). "Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia". Nature. 506 (7488): 328–333. doi:10.1038/nature13038. ISSN 1476-4687. PMC 4991939. PMID 24522528.
- Busque, Lambert; Patel, Jay P.; Figueroa, Maria E.; Vasanthakumar, Aparna; Provost, Sylvie; Hamilou, Zineb; Mollica, Luigina; Li, Juan; Viale, Agnes; Heguy, Adriana; Hassimi, Maryam; Socci, Nicholas; Bhatt, Parva K.; Gonen, Mithat; Mason, Christopher E.; Melnick, Ari; Godley, Lucy A.; Brennan, Cameron W.; Abdel-Wahab, Omar; Levine, Ross L. (1 November 2012). "Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis". Nature Genetics. 44 (11): 1179–1181. doi:10.1038/ng.2413. ISSN 1546-1718. PMC 3483435. PMID 23001125.
- Bureau, U.S. Census. "American FactFinder - Results". factfinder.census.gov. Archived from the original on 14 February 2020. Retrieved 1 May 2017.
- Young, Andrew L.; Challen, Grant A.; Birmann, Brenda M.; Druley, Todd E. (22 August 2016). "Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults". Nature Communications. 7: 12484. doi:10.1038/ncomms12484. ISSN 2041-1723. PMC 4996934. PMID 27546487.
- Abkowitz, Janis L.; Catlin, Sandra N.; McCallie, Monica T.; Guttorp, Peter (1 October 2002). "Evidence that the number of hematopoietic stem cells per animal is conserved in mammals". Blood. 100 (7): 2665–2667. doi:10.1182/blood-2002-03-0822. ISSN 0006-4971. PMID 12239184.
- Welch, John S.; Ley, Timothy J.; Link, Daniel C.; Miller, Christopher A.; Larson, David E.; Koboldt, Daniel C.; Wartman, Lukas D.; Lamprecht, Tamara L.; Liu, Fulu; Xia, Jun; Kandoth, Cyriac; Fulton, Robert S.; McLellan, Michael D.; Dooling, David J.; Wallis, John W.; Chen, Ken; Harris, Christopher C.; Schmidt, Heather K.; Kalicki-Veizer, Joelle M.; Lu, Charles; Zhang, Qunyuan; Lin, Ling; O'Laughlin, Michelle D.; McMichael, Joshua F.; Delehaunty, Kim D.; Fulton, Lucinda A.; Magrini, Vincent J.; McGrath, Sean D.; Demeter, Ryan T.; Vickery, Tammi L.; Hundal, Jasreet; Cook, Lisa L.; Swift, Gary W.; Reed, Jerry P.; Alldredge, Patricia A.; Wylie, Todd N.; Walker, Jason R.; Watson, Mark A.; Heath, Sharon E.; Shannon, William D.; Varghese, Nobish; Nagarajan, Rakesh; Payton, Jacqueline E.; Baty, Jack D.; Kulkarni, Shashikant; Klco, Jeffery M.; Tomasson, Michael H.; Westervelt, Peter; Walter, Matthew J.; Graubert, Timothy A.; DiPersio, John F.; Ding, Li; Mardis, Elaine R.; Wilson, Richard K. (20 July 2012). "The origin and evolution of mutations in acute myeloid leukemia". Cell. 150 (2): 264–278. doi:10.1016/j.cell.2012.06.023. ISSN 1097-4172. PMC 3407563. PMID 22817890.
- Klein, Allon M.; Simons, Benjamin D. (2011-08-01). "Universal patterns of stem cell fate in cycling adult tissues". Development. 138 (15): 3103–3111. doi:10.1242/dev.060103. ISSN 0950-1991. PMID 21750026.
- Heuser, Michael; Thol, Felicitas; Ganser, Arnold (6 May 2016). "Clonal Hematopoiesis of Indeterminate Potential". Deutsches Ärzteblatt International. 113 (18): 317–322. doi:10.3238/arztebl.2016.0317. ISSN 1866-0452. PMC 4961884. PMID 27215596.
- McKerrell, Thomas; Park, Naomi; Moreno, Thaidy; Grove, Carolyn S.; Ponstingl, Hannes; Stephens, Jonathan; Crawley, Charles; Craig, Jenny; Scott, Mike A.; Hodkinson, Clare; Baxter, Joanna; Rad, Roland; Forsyth, Duncan R.; Quail, Michael A.; Zeggini, Eleftheria; Ouwehand, Willem; Varela, Ignacio; Vassiliou, George S. (3 March 2015). "Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis". Cell Reports. 10 (8): 1239–1245. doi:10.1016/j.celrep.2015.02.005. ISSN 2211-1247. PMC 4542313. PMID 25732814.
- Montalban-Bravo, Guillermo; Garcia-Manero, Guillermo; List, Alan; Kantarjian, Hagop M.; Cortes, Jorge E. (1 June 2016). "Myelodysplastic Syndromes". www.cancernetwork.com.
- NIH SEER Program. "Acute Myeloid Leukemia (AML) Number of New Cases and Deaths Per 100,000 People (All Races, Males and Females), Age-Adjusted". NIH Surveillance, Epidemiology, and End Results Program (SEER). Archived from the original on 2017-02-03. Retrieved 2017-05-01.
- Fuster, José J.; MacLauchlan, Susan; Zuriaga, María A.; Polackal, Maya N.; Ostriker, Allison C.; Chakraborty, Raja; Wu, Chia-Ling; Sano, Soichi; Muralidharan, Sujatha; Rius, Cristina; Vuong, Jacqueline; Jacob, Sophia; Muralidhar, Varsha; Robertson, Avril A. B.; Cooper, Matthew A.; Andrés, Vicente; Hirschi, Karen K.; Martin, Kathleen A.; Walsh, Kenneth (24 February 2017). "Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice". Science. 355 (6327): 842–847. doi:10.1126/science.aag1381. ISSN 1095-9203. PMC 5542057. PMID 28104796.
- Zhu, Yanfang Peipei; Hedrick, Catherine C.; Gaddis, Dalia E. (24 February 2017). "Hematopoietic stem cells gone rogue". Science. 355 (6327): 798–799. doi:10.1126/science.aam7939. ISSN 1095-9203. PMID 28232539.
- Tall, Alan R.; Levine, Ross L. (2 March 2017). "Cardiovascular disease: Commonality with cancer". Nature. 543 (7643): 45–47. doi:10.1038/nature21505. ISSN 1476-4687. PMID 28225756.