Chimeric antigen receptor T cell
Chimeric antigen receptor T cells (also known as CAR T cells) are T cells that have been genetically engineered to produce an artificial T-cell receptor for use in immunotherapy.[1]
Chimeric antigen receptors (CARs, also known as chimeric immunoreceptors, chimeric T cell receptors or artificial T cell receptors) are receptor proteins that have been engineered to give T cells the new ability to target a specific protein. The receptors are chimeric because they combine both antigen-binding and T-cell activating functions into a single receptor.
CAR-T cell therapy uses T cells engineered with CARs for cancer therapy. The premise of CAR-T immunotherapy is to modify T cells to recognize cancer cells in order to more effectively target and destroy them. Scientists harvest T cells from people, genetically alter them, then infuse the resulting CAR-T cells into patients to attack their tumors.[2] CAR-T cells can be either derived from T cells in a patient's own blood (autologous) or derived from the T cells of another healthy donor (allogeneic). Once isolated from a person, these T cells are genetically engineered to express a specific CAR, which programs them to target an antigen that is present on the surface of tumors. For safety, CAR-T cells are engineered to be specific to an antigen expressed on a tumor that is not expressed on healthy cells.[3]
After CAR-T cells are infused into a patient, they act as a "living drug" against cancer cells.[4] When they come in contact with their targeted antigen on a cell, CAR-T cells bind to it and become activated, then proceed to proliferate and become cytotoxic.[5] CAR-T cells destroy cells through several mechanisms, including extensive stimulated cell proliferation, increasing the degree to which they are toxic to other living cells (cytotoxicity) and by causing the increased secretion of factors that can affect other cells such as cytokines, interleukins and growth factors.[6]
Production
The first step in the production of CAR-T cells is the isolation of T cells from human blood. CAR-T cells may be manufactured either from the patient's own blood, known as an autologous treatment, or from the blood of a healthy donor, known as an allogeneic treatment. The manufacturing process is the same in both cases; only the choice of initial blood donor is different.
First, leukocytes are isolated using a blood cell separator in a process known as leukocyte apheresis. Peripheral blood mononuclear cells (PBMC) are then separated and collected.[7] The products of leukocyte apheresis are then transferred to a cell-processing center. In the cell processing center, specific T cells are stimulated so that they will actively proliferate and expand to large numbers. To drive their expansion, T cells are typically treated with the cytokine interleukin 2 (IL-2) and anti-CD3 antibodies.[8]
The expanded T cells are purified and then transduced with a gene encoding the engineered CAR via a retroviral vector, typically either an integrating gammaretrovirus (RV) or a lentiviral (LV) vector. These vectors are very safe in modern times due to a partial deletion of the U3 region.[9] The new gene editing tool CRISPR/Cas9 has recently been used instead of retroviral vectors to integrate the CAR gene into specific sites in the genome.[1]
The patient undergoes lymphodepletion chemotherapy prior to the introduction of the engineered CAR-T cells.[10] The depletion of the number of circulating leukocytes in the patient upregulates the number of cytokines that are produced and reduces competition for resources, which helps to promote the expansion of the engineered CAR-T cells.[11]
Cancer treatment
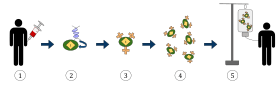
1. T-cells (represented by objects labeled as ’t’) are removed from the patient's blood.
2. Then in a lab setting the gene that encodes for the specific antigen receptors are incorporated into the T-cells.
3. Thus producing the CAR receptors (labeled as c) on the surface of the cells.
4. The newly modified T-cells are then further harvested and grown in the lab.
5. After a certain time period, the engineered T-cells are infused back into the patient.
T cells are genetically engineered to express chimeric antigen receptors specifically directed toward antigens on a patient's tumor cells, then infused into the patient where they attack and kill the cancer cells.[12] Adoptive transfer of T cells expressing CARs is a promising anti-cancer therapeutic, because CAR-modified T cells can be engineered to target virtually any tumor associated antigen.
Early CAR-T cell research has focused on blood cancers. The first approved treatments use CARs that target the antigen CD19, present in B-cell-derived cancers such as acute lymphoblastic leukemia (ALL) and diffuse large B-cell lymphoma (DLBCL).[13] [14] There are also efforts underway to engineer CARs targeting many other blood cancer antigens, including CD30 in refractory Hodgkin's lymphoma; CD33, CD123, and FLT3 in acute myeloid leukemia (AML); and BCMA in multiple myeloma.[15]
Solid tumors have presented a more difficult target.[16] Identification of good antigens has been challenging: such antigens must be highly expressed on the majority of cancer cells, but largely absent on normal tissues.[17] CAR-T cells are also not trafficked efficiently into the center of solid tumor masses, and the hostile tumor microenvironment suppresses T cell activity.[15]
Clinical studies and FDA approvals
The first two FDA-approved CAR-T therapies both target the CD19 antigen, which is found on many types of B-cell cancers.[18] Tisagenlecleucel (Kymriah / Novartis) is approved to treat relapsed/refractory B-cell precursor acute lymphoblastic leukemia (ALL), while axicabtagene ciloleucel (Yescarta / Kite Pharma) is approved to treat relapsed/refractory diffuse large B-cell lymphoma (DLBCL).[18]
As of March 2019, there were around 364 ongoing clinical trials happening globally involving CAR-T cells.[19] The majority of those trials target blood cancers: CAR-T therapies account for more than half of all trials for hematological malignancies.[19] CD19 continues to be the most popular antigen target,[20] followed by BCMA (commonly expressed in multiple myeloma).[19][21] In 2016, studies began to explore the viability of other antigens, such as CD20.[22] Trials for solid tumors are less dominated by CAR-T, with about half of cell therapy-based trials involving other platforms such as NK cells.[19]
Although the initial clinical remission rates after CAR-T cell therapy in all patients are as high as 90%,[23] long term survival rates are much lower. The cause is typically the emergence of leukemia cells that do not express CD19 and so evade recognition by the CD19–CAR T cells, a phenomenon known as antigen escape.[15] Preclinical studies developing CAR-T cells with dual targeting of CD19 plus CD22 or CD19 plus CD20 have demonstrated promise, and trials studying bispecific targeting to circumvent CD19 down-regulation are ongoing.[15]
Safety concerns
There are serious side effects that result from CAR-T cells being introduced into the body, including cytokine release syndrome and neurological toxicity.[10] Because it is a relatively new treatment, there is little data about the long-term effects of CAR-T cell therapy. There are still concerns about long-term patient survival, as well as pregnancy complications in female patients treated with CAR-T cells.[24]
The most common issue after treatment with CAR-T cells is cytokine release syndrome (CRS), a condition in which the immune system is activated and releases an increased number of inflammatory cytokines. The clinical manifestation of this syndrome resembles sepsis with high fever, fatigue, myalgia, nausea, capillary leakages, tachycardia and other cardiac dysfunction, liver failure, and kidney impairment.[25] CRS occurs in almost all patients treated with CAR-T cell therapy; in fact, the presence of CRS is a diagnostic marker that indicates the CAR-T cells are working as intended to kill the cancer cells.[24] Note, however, that a higher grade of CRS severity does not correlate with an increased response to the treatment, but rather higher disease burden.[24]
Neurological toxicity is also often associated with CAR-T cell treatment.[26] The underlying mechanism is poorly understood, and may or may not be related to CRS. Clinical manifestations include delirium, the partial loss of the ability to speak coherently while still having the ability to interpret language (expressive aphasia), lowered alertness (obtundation), and seizures.[24] During some clinical trials deaths caused by neurotoxicity have occurred. The main cause of death from neurotoxicity is cerebral edema. In a study carried out by Juno Therapeutics, Inc., five patients enrolled in the trial died as a result of cerebral edema. Two of the patients were treated with cyclophosphamide alone and the remaining three were treated with a combination of cyclophosphamide and fludarabine.[27] In another clinical trial sponsored by the Fred Hutchinson Cancer Research Center, there was one reported case of irreversible and fatal neurological toxicity 122 days after the administration of CAR-T cells.[28]
Anaphylaxis is an expected side effect, as the CAR is made with a foreign monoclonal antibody and as a result, provokes an immune response.
On-target/off-tumor recognition occurs when the CAR-T cell recognizes the correct antigen, but the antigen is expressed on healthy, non-pathogenic tissue. This results in the CAR-T cells attacking non-tumor tissue, such as healthy B cells that express CD19. The severity of this adverse effect can vary from B-cell aplasia, which can be treated with supporting infusions, to extreme toxicity leading to death.[8]
There is also the unlikely possibility that the engineered CAR-T cells will themselves become transformed into cancerous cells through insertional mutagenesis, due to the viral vector inserting the CAR gene into a tumor suppressor or oncogene in the host T cell's genome. Some retroviral (RV) vectors carry a lower risk than lentiviral (LV) vectors. However, both have the potential to be oncogenic. Genomic sequencing analysis of CAR insertion sites in T cells has been established for better understanding of CAR T cell function and persistence in vivo.[17]
Receptor structure
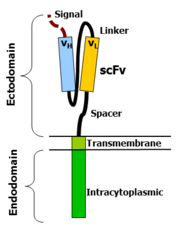
Chimeric antigen receptors combine many facets of normal T cell activation into a single protein. They link an extracellular antigen recognition domain to an intracellular signalling domain, which activates the T cell when an antigen is bound. CARs are composed of four regions: an antigen recognition domain, an extracellular hinge region, a transmembrane domain, and an intracellular T-cell signaling domain.[29][30]
Antigen recognition domain
The antigen recognition domain is exposed to the outside of the cell, in the ectodomain portion of the receptor. It interacts with potential target molecules and is responsible for targeting the CAR-T cell to any cell expressing a matching molecule.
The antigen recognition domain is typically derived from the variable regions of a monoclonal antibody linked together as a single-chain variable fragment (scFv).[30] An scFv is a chimeric protein made up of the light (VL) and heavy (VH) chains of immunoglobins, connected with a short linker peptide.[31] These VL and VH regions are selected in advance for their binding ability to the target antigen (such as CD19). The linker between the two chains consists of hydrophilic residues with stretches of glycine and serine in it for flexibility as well as stretches of glutamate and lysine for added solubility.[32] Single domain antibodies (e.g. VH, VHH) have been engineered and developed as antigen recognition domains in the CAR format due to their high transduction efficiency in T cells. [33][17]
In addition to antibody fragments, non‐antibody‐based approaches have also been used to direct CAR specificity, usually taking advantage of ligand/receptor pairs that normally bind to each other.[29] Cytokines, innate immune receptors, TNF receptors, growth factors, and structural proteins have all been successfully used as CAR antigen recognition domains.[29]
Hinge region
The hinge, also called a spacer, is a small structural domain that sits between the antigen recognition region and the cell's outer membrane. An ideal hinge enhances the flexibility of the scFv receptor head, reducing the spatial constraints between the CAR and its target antigen. This promotes antigen binding and synapse formation between the CAR-T cells and target cells.[34] Hinge sequences are often based on membrane-proximal regions from other immune molecules including IgG, CD8, and CD28.[29][35]
Transmembrane domain
The transmembrane domain is a structural component, consisting of a hydrophobic alpha helix that spans the cell membrane. It anchors the CAR to the plasma membrane, bridging the extracellular hinge and antigen recognition domains with the intracellular signaling region.[29] This domain is essential for the stability of the receptor as a whole. Generally, the transmembrane domain from the most membrane-proximal component of the endodomain is used, but different transmembrane domains result in different receptor stability. The CD28 transmembrane domain is known to result in a highly expressed, stable receptor.
Using the CD3-zeta transmembrane domain is not recommended, as it can result in incorporation of the artificial TCR into the native TCR.[36]
Intracellular T-cell signaling domain
The intracellular T-cell signaling domain lies in the receptor's endodomain, inside the cell.[29] After an antigen is bound to the external antigen recognition domain, CAR receptors cluster together and transmit an activation signal. Then the internal cytoplasmic end of the receptor perpetuates signaling inside the T cell.[31]
Normal T cell activation relies on the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) present in the cytoplasmic domain of CD3-zeta. To mimic this process, CD3-zeta's cytoplasmic domain is commonly used as the main CAR endodomain component. Other ITAM-containing domains have also been tried, but are not as effective.[30]
T cells also require co-stimulatory molecules in addition to CD3 signaling in order to persist after activation. For this reason, the endodomains of CAR receptors typically also include one or more chimeric domains from co-stimulatory proteins.[37] Signaling domains from a wide variety of co-stimulatory molecules have been successfully tested, including CD28, CD27, CD134 (OX40), and CD137 (4‐1BB).[29]
Evolution of CAR design
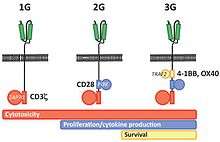
The first CAR-T cells were developed in 1987 by Yoshikazu Kuwana et al.[39] followed in 1989 by Gideon Gross and Zelig Eshhar[40][41] at Weizmann Institute, Israel.[42] The sophistication of the engineered CAR receptors has grown over time, and are referred to as first, second, third, or fourth generation CARs depending on their composition.
First generation CARs are composed of an extracellular binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains.[5] The extracellular binding domain contains a single-chain variable fragment (scFv) derived from tumor antigen‐reactive antibodies that usually has a high specificity to a specific tumor antigen.[5] All CARs contain the CD3ζ chain domain as the intracellular signaling domain, which is the primary transmitter of T cell activation signals.
Second generation CARs add a co‐stimulatory domain, like CD28 or 4‐1BB. The involvement of these intracellular signaling domains improve T cell proliferation, cytokine secretion, resistance to apoptosis, and in vivo persistence.[5]
Third generation CARs combine multiple co-stimulatory domains, such as CD28-41BB or CD28-OX40, to augment T cell activity. Preclinical data show the third-generation CARs exhibit improved effector functions and better in vivo persistence as compared to second‐generation CARs.[5]
Fourth generation CARs (also known as TRUCKs or armored CARs) further add factors that enhance T cell expansion, persistence, and anti‐tumoral activity. This can include cytokines, such is IL-2, IL-5, IL-12 and co‐stimulatory ligands.[43][44]
in 2018, a version of CAR was developed that is referred to as SUPRA CAR, or split, universal, and programmable.[45] Multiple mechanisms can be deployed to finely regulate the activity of SUPRA CAR, which limits overactivation. In contrast to the traditional CAR design, SUPRA CAR allows targeting of multiple antigens without further genetic modification of a person's immune cells.[46]
Control mechanisms
Adding a synthetic control mechanism to engineered T cells allows doctors to precisely control the persistence or activity of the T cells in the patient's body, with the goal of reducing toxic side effects.[47] The major control techniques trigger T cell death or limit T cell activation, and often regulate the T cells via a separate drug that can be introduced or withheld as needed.
Suicide genes: Genetically modified T cells are engineered to include one or more genes that can induce apoptosis when activated by an extracellular molecule. Herpes simplex virus thymidine kinase (HSV-TK) and inducible caspase 9 (iCasp9) are two types of suicide genes that have been integrated into CAR-T cells.[47][48][49] In the iCasp9 system, the suicide gene complex has two elements: a mutated FK506-binding protein with high specificity to the small molecule rimiducid/AP1903, and a gene encoding a pro-domain-deleted human caspase 9. Dosing the patient with rimiducid activates the suicide system, leading to rapid apoptosis of the genetically modified T cells. Although both the HSV-TK and iCasp9 systems demonstrate a noticeable function as a safety switch in clinical trials, some defects limit their application. HSV-TK is virus-derived and may be immunogenic to humans.[47][50] It is also currently unclear whether the suicide gene strategies will act quickly enough in all situations to halt dangerous off-tumor cytotoxicity.
Dual-antigen receptor: CAR-T cells are engineered to express two tumor-associated antigen receptors at the same time, reducing the likelihood that the T cells will attack non-tumor cells. Dual-antigen receptor CAR-T cells have been reported to have less intense side effects.[51] An in vivo study in mice shows that dual-receptor CAR-T cells effectively eradicated prostate cancer and achieved complete long-term survival.[52]
ON-switch: In this system, CAR-T cells can only function in the presence of both tumor antigen and a benign exogenous molecule. To achieve this, the CAR-T cell's engineered chimeric antigen receptor is split into two separate proteins that must come together in order to function. The first receptor protein typically contains the extracellular antigen binding domain, while the second protein contains the downstream signaling elements and co-stimulatory molecules (such as CD3ζ and 4-1BB). In the presence of an exogenous molecule (such as a rapamycin analog), the binding and signaling proteins dimerize together, allowing the CAR-T cells to attack the tumor.[53]
Bispecific molecules as switches: Bispecific molecules target both a tumor-associated antigen and the CD3 molecule on the surface of T cells. This ensures that the T cells cannot become activated unless they are in close physical proximity to a tumor cell.[54] The anti-CD20/CD3 bispecific molecule shows high specificity to both malignant B cells and cancer cells in mice.[55] FITC is another bifunctional molecule used in this strategy. FITC can redirect and regulate the activity of the FITC-specific CAR-T cells toward tumor cells with folate receptors.[56]
Small molecule drug conjugates adaptor technology
SMDCs (small molecule drug conjugates) platform in immuno-oncology is an experimental approach that makes possible the engineering of a single universal CAR T cell, which binds with extraordinarily high affinity to a benign molecule designated as fluorescein isothiocyanate (FITC). These cells are then used to treat various cancer types when co-administered with bispecific SMDC adaptor molecules. These unique bispecific adaptors are constructed with a FITC molecule and a tumor-homing molecule to precisely bridge the universal CAR T cell with the cancer cells, which causes localized T cell activation. Anti-tumor activity in mice is induced only when both the universal CAR T cells plus the correct antigen-specific adaptor molecules are present. Anti-tumor activity and toxicity can be controlled by adjusting the administered adaptor molecule dosing. Treatment of antigenically heterogeneous tumors can be achieved by administration of a mixture of the desired antigen-specific adaptors.
There are several challenges of current CAR T cell therapies, such as:
- the inability to control the rate of cytokine release and tumor lysis
- the absence of an off switch that would terminate cytotoxic activity when tumor eradication is complete
- a requirement to generate a different CAR T cell for each unique tumor antigen may be solved or mitigated using the adaptor approach.[57][58]
See also
References
- Jensen TI, Axelgaard E, Bak RO (June 2019). "Therapeutic gene editing in haematological disorders with CRISPR/Cas9". British Journal of Haematology. 185 (5): 821–835. doi:10.1111/bjh.15851. PMID 30864164.
- Fox M (July 12, 2017). "New Gene Therapy for Cancer Offers Hope to Those With No Options Left". NBC News.
- Srivastava S, Riddell SR (August 2015). "Engineering CAR-T cells: Design concepts". Trends in Immunology. 36 (8): 494–502. doi:10.1016/j.it.2015.06.004. PMC 4746114. PMID 26169254.
- Sadelain M, Brentjens R, Rivière I (April 2013). "The basic principles of chimeric antigen receptor design". Cancer Discovery. 3 (4): 388–98. doi:10.1158/2159-8290.CD-12-0548. PMC 3667586. PMID 23550147.
- Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ (2017). "Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts". EMBO Molecular Medicine. 9 (9): 1183–1197. doi:10.15252/emmm.201607485. PMC 5582407. PMID 28765140.
- Tang XJ, Sun XY, Huang KM, Zhang L, Yang ZS, Zou DD, Wang B, Warnock GL, Dai LJ, Luo J (December 2015). "Therapeutic potential of CAR-T cell-derived exosomes: a cell-free modality for targeted cancer therapy". Oncotarget. 6 (42): 44179–90. doi:10.18632/oncotarget.6175. PMC 4792550. PMID 26496034.
- Jin C, Yu D, Hillerdal V, Wallgren A, Karlsson-Parra A, Essand M (2014-03-05). "Allogeneic lymphocyte-licensed DCs expand T cells with improved antitumor activity and resistance to oxidative stress and immunosuppressive factors". Molecular Therapy. Methods & Clinical Development. 1: 14001. doi:10.1038/mtm.2014.1. PMC 4362340. PMID 26015949.
- Makita S, Yoshimura K, Tobinai K (June 2017). "Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma". Cancer Science. 108 (6): 1109–1118. doi:10.1111/cas.13239. PMC 5480083. PMID 28301076.
- Jin C, Fotaki G, Ramachandran M, Nilsson B, Essand M, Yu D (July 2016). "Safe engineering of CAR T cells for adoptive cell therapy of cancer using long-term episomal gene transfer". EMBO Molecular Medicine. 8 (7): 702–11. doi:10.15252/emmm.201505869. PMC 4931286. PMID 27189167.
- Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ (September 2017). "Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts". EMBO Molecular Medicine. 9 (9): 1183–1197. doi:10.15252/emmm.201607485. PMC 5582407. PMID 28765140.
- Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, Restifo NP (December 2006). "Increased intensity lymphodepletion and adoptive immunotherapy--how far can we go?". Nature Clinical Practice Oncology. 3 (12): 668–81. doi:10.1038/ncponc0666. PMC 1773008. PMID 17139318.
- Jacobson CA, Ritz J (November 2011). "Time to put the CAR-T before the horse". Blood. 118 (18): 4761–2. doi:10.1182/blood-2011-09-376137. PMID 22053170.
- Kochenderfer, et al. (2010). "Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. 2010". Blood. 116 (20): 4099–102. doi:10.1182/blood-2010-04-281931. PMC 2993617. PMID 20668228.
- Kochenderfer, et al. (2015). "Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor". Journal of Clinical Oncology. 2015 (33): 540–9. doi:10.1200/JCO.2014.56.2025. PMC 4322257. PMID 25154820.
- Schultz L, Mackall C (2019). "Driving CAR T cell translation forward". Science Translational Medicine. 11 (481): eaaw2127. doi:10.1126/scitranslmed.aaw2127. PMID 30814337.
- Lim WA, June CH (February 2017). "The Principles of Engineering Immune Cells to Treat Cancer". Cell. 168 (4): 724–740. doi:10.1016/j.cell.2017.01.016. PMC 5553442. PMID 28187291.
- Li D, Li N, Zhang YF, Fu H, Feng M, Schneider D, et al. (February 2020). "Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice". Gastroenterology. 158 (8): 2250–2265.e20. doi:10.1053/j.gastro.2020.02.011. PMID 32060001.
- "Thumbs Up to Latest CAR T-Cell Approval". www.medpagetoday.com. 2017-10-19. Retrieved 2019-04-19.
- Yu JX, Hubbard-Lucey VM, Tang J (2019-05-30). "The global pipeline of cell therapies for cancer". Nature Reviews Drug Discovery. 18 (11): 821–822. doi:10.1038/d41573-019-00090-z. PMID 31673124. S2CID 190862546.
- Brudno and Kochenderfer. Chimeric antigen receptor T-cell therapies for lymphoma. Nature Reviews Clinical Oncology. 2018. 15: 31-46.
- Mikkilineni and Kochenderfer. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. 2017. 130: 2594-602
- Almåsbak H, Aarvak T, Vemuri MC (2016). "CAR T Cell Therapy: A Game Changer in Cancer Treatment". Journal of Immunology Research. 2016: 5474602. doi:10.1155/2016/5474602. PMC 4889848. PMID 27298832.
- "A Cure for Cancer? How CAR-T Therapy is Revolutionizing Oncology" (Press release). labiotech. March 8, 2018. Retrieved April 19, 2018.
- Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ (2016). "Toxicity and management in CAR-T cell therapy". Molecular Therapy Oncolytics. 3: 16011. doi:10.1038/mto.2016.11. PMC 5008265. PMID 27626062.
- Breslin S (February 2007). "Cytokine-release syndrome: overview and nursing implications". Clinical Journal of Oncology Nursing. 11 (1 Suppl): 37–42. doi:10.1188/07.CJON.S1.37-42. PMID 17471824. S2CID 35773028.
- Brudno JN, Kochenderfer JN (June 2016). "Toxicities of chimeric antigen receptor T cells: recognition and management". Blood. 127 (26): 3321–3330. doi:10.1182/blood-2016-04-703751. PMC 4929924. PMID 27207799.
- "Study Evaluating the Efficacy and Safety of JCAR015 in Adult B-cell Acute Lymphoblastic Leukemia (B-ALL)". ClinicalTrials.gov. Retrieved 2018-02-21.
- Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG (June 2016). "CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients". The Journal of Clinical Investigation. 126 (6): 2123–38. doi:10.1172/JCI85309. PMC 4887159. PMID 27111235.
- Chandran SS, Klebanoff CA (9 May 2019). "T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance". Immunological Reviews. 290 (1): 127–147. doi:10.1111/imr.12772. PMC 7027847. PMID 31355495.

- Dotti G, Gottschalk S, Savoldo B, Brenner MK (January 2014). "Design and development of therapies using chimeric antigen receptor-expressing T cells". Immunological Reviews. 257 (1): 107–26. doi:10.1111/imr.12131. PMC 3874724. PMID 24329793.
- Zhang C, Liu J, Zhong JF, Zhang X (2017-06-24). "Engineering CAR-T cells". Biomarker Research. 5: 22. doi:10.1186/s40364-017-0102-y. PMC 5482931. PMID 28652918.
- Baldo BA (May 2015). "Chimeric fusion proteins used for therapy: indications, mechanisms, and safety". Drug Safety. 38 (5): 455–79. doi:10.1007/s40264-015-0285-9. PMID 25832756. S2CID 23852865.
- Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M (August 2017). "Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma". Proceedings of the National Academy of Sciences of the United States of America. 114 (32): E6623–E6631. doi:10.1073/pnas.1706055114. PMC 5559039. PMID 28739923.
- Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR (Feb 2015). "The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity". Cancer Immunology Research. 3 (2): 125–35. doi:10.1158/2326-6066.CIR-14-0127. PMC 4692801. PMID 25212991.
- Qin L, Lai Y, Zhao R, Wei X, Weng J, Lai P, Li B, Lin S, Wang S, Wu Q, Liang Q, Li Y, Zhang X, Wu Y, Liu P, Yao Y, Pei D, Du X, Li P (March 2017). "Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells". Journal of Hematological Oncology. 10 (1): 68. doi:10.1186/s13045-017-0437-8. PMC 5347831. PMID 28288656.
- Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE (June 2010). "The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex". Journal of Immunology. 184 (12): 6938–49. doi:10.4049/jimmunol.0901766. PMID 20483753.
- Sadelain M, Brentjens R, Riviere I (April 2013). "The basic principles of chimeric antigen receptor (CAR) design". Cancer Discovery. 3 (4): 388–398. doi:10.1158/2159-8290.CD-12-0548. PMC 3667586. PMID 23550147.
- Casucci M, Bondanza A (2011). "Suicide gene therapy to increase the safety of chimeric antigen receptor-redirected T lymphocytes". Journal of Cancer. 2: 378–82. doi:10.7150/jca.2.378. PMC 3133962. PMID 21750689.
- Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y (February 1989). "Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions". Biochem Biophys Res Commun. 149 (3): 960–968. doi:10.1016/0006-291x(87)90502-x. PMID 3122749.
- Gross G, Gorochov G, Waks T, Eshhar Z (February 1989). "Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity". Transplantation Proceedings. 21 (1 Pt 1): 127–30. PMID 2784887.
- Rosenbaum L (October 2017). "Tragedy, Perseverance, and Chance - The Story of CAR-T Therapy". The New England Journal of Medicine. 377 (14): 1313–1315. doi:10.1056/NEJMp1711886. PMID 28902570.
- Gross G, Waks T, Eshhar Z (December 1989). "Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity". Proceedings of the National Academy of Sciences of the United States of America. 86 (24): 10024–8. Bibcode:1989PNAS...8610024G. doi:10.1073/pnas.86.24.10024. JSTOR 34790. PMC 298636. PMID 2513569.
- Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE (March 2018). "CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity". Molecular Therapy Oncolytics. 8: 41–51. doi:10.1016/j.omto.2017.12.003. PMC 5772011. PMID 29367945.
- Chmielewski M, Abken H (2015). "TRUCKs: the fourth generation of CARs". Expert Opinion on Biological Therapy. 15 (8): 1145–54. doi:10.1517/14712598.2015.1046430. PMID 25985798.
- Choe, Joseph H.; Williams, Jasper Z.; Lim, Wendell A. (2020). "Engineering T Cells to Treat Cancer: The Convergence of Immuno-Oncology and Synthetic Biology". Annual Review of Cancer Biology. 4: 121–139. doi:10.1146/annurev-cancerbio-030419-033657.
- Cho, Jang Hwan; Collins, James J.; Wong, Wilson W. (2018). "Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses". Cell. 173 (6): 1426–1438.e11. doi:10.1016/j.cell.2018.03.038. PMC 5984158. PMID 29706540.
- Zhang E, Xu H (2017). "A new insight in chimeric antigen receptor‐engineered T cells for cancer immunotherapy". Hematol Oncol. 10 (1): 1. doi:10.1186/s13045-016-0379-6. PMC 5210295. PMID 28049484.
- Bonini C, Ferrari G (1997). "HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia". Science. 276 (5319): 1719–1724. doi:10.1126/science.276.5319.1719. PMID 9180086.
- Quintarelli C, Vera JF (2007). "Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes". Blood. 110 (8): 2793–2802. doi:10.1182/blood-2007-02-072843. PMC 2018664. PMID 17638856.
- Riddell SR, Elliott M (1996). "T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients". Nat Med. 2 (2): 216–223. doi:10.1038/nm0296-216. PMID 8574968. S2CID 35503876.
- Maher J, Brentjens RJ (2002). "Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor". Nat Biotechnol. 20 (1): 70–75. doi:10.1038/nbt0102-70. PMID 11753365.
- Wilkie S, van Schalkwyk MC (2012). "Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling". Clin Immunol. 32 (5): 1059–1070. doi:10.1007/s10875-012-9689-9. PMID 22526592. S2CID 17660404.
- Wu CY (2015). "Remote control of therapeutic T cells through a small molecule-gated chimeric receptor". Science. 350 (6258): aab4077. Bibcode:2015Sci...350.4077W. doi:10.1126/science.aab4077. PMC 4721629. PMID 26405231.
- Frankel SR (2013). "Targeting T cells to tumor cells using bispecific antibodies". Curr Opin Chem Biol. 17 (3): 385–392. doi:10.1016/j.cbpa.2013.03.029. PMID 23623807.
- Sun LL (2015). "Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies". Sci Transl Med. 7 (287): 287ra70. doi:10.1126/scitranslmed.aaa4802. PMID 25972002. S2CID 24939667.
- Kim CH (2013). "Bispecific small molecule-antibody conjugate targeting prostate cancer". PNAS. 110 (44): 17796–17801. Bibcode:2013PNAS..11017796K. doi:10.1073/pnas.1316026110. PMC 3816437. PMID 24127589.
- SMDC technology. Archived 2016-03-27 at the Wayback Machine ENDOCYTE
- "Endocyte announces promising preclinical data for application of SMDC technology in CAR T cell therapy in late-breaking abstract at American Association for Cancer Research (AACR) annual meeting 2016" (Press release). Endocyte. April 19, 2016. Archived from the original on July 30, 2017. Retrieved December 20, 2017.