Bite angle
In coordination chemistry the bite angle is the ligand–metal–ligand bond angle of coordination complex containing a bidentate ligand. This geometric parameter is used to classify chelating ligands, including those in organometallic complexes. It is most often discussed in terms of catalysis, as changes in bite angle can affect not just the activity and selectivity of a catalytic reaction but even allow alternative reaction pathways to become accessible.[1][2][3]
-3D-balls.png)
Although the parameter can be applied generally to any chelating ligand, it is commonly applied to describe diphosphine ligands, as they can adopt a wide range of bite angles.[2][3]
Diamines
Diamines form a wide range of coordination complexes. They typically form 5- and 6-membered chelate rings. Examples of the former include ethylenediamine and 2,2′-bipyridine. Six-membered chelate rings are formed by 1,3-diaminopropane. The bite angle in such complexes is usually near 90°. Longer chain diamines, which are "floppy", tend not to form chelate rings.[4]
Diphosphines
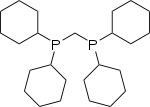
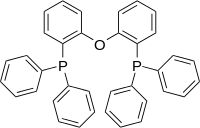
Diphosphines are a class of chelating ligands that contain two phosphine groups connected by a bridge (also referred to as a backbone). The bridge, for instance, might consist of one or more methylene groups or multiple aromatic rings with heteroatoms attached. Examples of common diphosphines are dppe, dcpm (Figure 1), and DPEphos (Figure 2). The structure of the backbone and the substituents attached to the phosphorus atoms influence the chemical reactivity of the diphosphine ligand in metal complexes through steric and electronic effects.[5]
Examples
Steric characteristics of the diphosphine ligand that influence the regioselectivity and rate of catalysis include the pocket angle, solid angle, repulsive energy, and accessible molecular surface.[6] Also of importance is the cone angle, which in diphosphines is defined as the average of the cone angle for the two substituents attached to the phosphorus atoms, the bisector of the P–M–P angle, and the angle between each M–P bond.[7] Larger cone angles usually result in faster dissociation of phosphine ligands because of steric crowding.
The natural bite angle
The natural bite angle (βn) of diphosphines, obtained using molecular mechanics calculations, is defined as the preferred chelation angle determined only by ligand backbone and not by metal valence angles (Figure 3).[1]

Both steric bite angle effect and the electronic bite angle effects are recognized.[7] The steric bite angle effect involves the steric interactions between ligands or between a ligand and a substrate. The electronic bite angle effect, on the other hand, relates to the electronic changes that occur when the bite angle is modified. This effect is sensitive to the hybridization of metal orbitals.[8] This flexibility range accounts for the diverse conformations of the ligand with energies slightly above the strain energy of the natural bite angle.
The bite angle of a diphosphine ligand also indicates the distortion from the ideal geometry of a complex based on VSEPR models. Octahedral and square planar complexes prefer angles near 90° while tetrahedral complexes prefer angles near 110°. Since catalysts often interconvert between various geometries, the rigidity of the chelate ring can be decisive.[8] A bidentate phosphine with a natural bite angle of 120° may preferentially occupy two equatorial sites in a trigonal bipyramidal complex whereas a bidentate phosphine with a natural bite angle of 90° may preferentially occupy apical-equatorial positions.[9] Diphosphine ligands with bite angles of over 120° are obtained using a bulky, stiff diphosphine backbones.[8] Diphosphines of wide bite angles are used in some industrial processes.
A case study: hydroformylation
The hydroformylation of alkenes to give aldehydes is an important industrial process. Almost 6 million tons of aldehydes are produced by this method annually.[9] Rhodium complexes containing diphosphine ligands are active hydroformylation catalysts. The ratio of linear to branched aldehyde product depends on the structure of the catalyst.[9][10]
One intermediate, [Rh(H)(alkene)(CO)L], exists in two different isomers, depending on the position of phosphine ligands (Figure 4).[9]
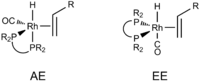
Diphosphine ligands such as dppe, which has a bite angle of about 90°, span the equatorial and apical positions (AE isomer). Diphosphines with larger bite angles (above 120°) preferentially occupy a pair of equatorial positions (EE isomer). It is believed that the EE isomer favors formation of linear aldehydes, the desired product. In an effort to create rhodium complexes in which the phosphine ligands preferentially occupy the equatorial positions, the use of diphosphine ligands with wide bite angles such as BISBI (Figure 5) has been investigated.
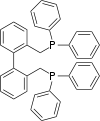 Figure 5. BISBI, a diphosphine with a bite angle of 113°.
Figure 5. BISBI, a diphosphine with a bite angle of 113°.
With a bite angle of approximately 113°, BISBI spans sites on equatorial plane of the trigonal bipyramidal intermediate complex (Figure 6).[8]
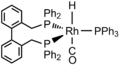
The structure of the intermediate [Rh(H)(diphosphine)(CO)2] does not however determine the regioselectivity of the hydroformylation. Instead, the formation of the linear vs. branched aldehydes is determined upon formation of [Rh(H)(diphosphine)CO(alkene)] and the subsequent hydride migration step. The bite angle affect the steric crowding at the Rh atom that results from the interactions of the bulky backbone of the ligand with substrate. The wide bite angle that results from the backbone allows the five-coordinate [Rh(H)(diphosphine)CO(alkene)] intermediate to adopt a structure that relieves steric hindrance. Thus, BISBI occupies the equatorial positions, where it has the most space. This preference of a transition state that relieves steric hindrance favors the formation of the linear aldehyde. The regioselectivity is also controlled by the hydride migration, which is usually irreversible in the formation of linear aldehydes.[8]
Furthermore, studies using Xantphos ligands (ligands with bulky backbones) in hydroformylation have indicated an increase in the rate of catalysis in metal complexes that contain diphosphine ligands with larger bite angles.[8] The electronic effect of this increase in reaction rate is uncertain since it mainly depends on the bonding between the alkene and rhodium.[9] Large bite angles promote alkene to rhodium electron donation, which results in an accumulation of electron density on the rhodium atom. This increased electron density would be available for π-donation into the anti-bonding orbitals of other ligands, which could weaken other M-L bonds within the catalyst, leading to higher rates.
The application of diphosphine ligands to catalysts is not limited to the process of hydroformylation. Hydrocyanation and hydrogenation reactions also implement phosphine-mediated catalysts.
References
- van Leeuwen, P. W. N. M.; Kamer, P. C. J.; Reek, J. N. H. (30 August 1999). "The bite angle makes the catalyst". Pure and Applied Chemistry. 71 (8): 1443–1452. doi:10.1351/pac199971081443.

- Dierkes, Peter; van Leeuwen, Piet W. N. M. (1999). "The bite angle makes the difference: a practical ligand parameter for diphosphine ligands". Journal of the Chemical Society, Dalton Transactions (10): 1519–1530. doi:10.1039/A807799A.
- Birkholz (née Gensow), Mandy-Nicole; Freixa, Zoraida; van Leeuwen, Piet W. N. M. (2009). "Bite angle effects of diphosphines in C–C and C–X bond forming cross coupling reactions". Chemical Society Reviews. 38 (4): 1099–118. doi:10.1039/B806211K. PMID 19421583.
- Zelewsky, A. von (1995). Stereochemistry of Coordination Compounds. Chichester: John Wiley. ISBN 047195599X.
- Iwamoto, M.; Yuguchi, S. (1966). "Reaction of Butadiene with Ethylene. II. New Catalytic Systems in Synthesis of 1,4-Hexadiene". J. Org. Chem. 31 (12): 4290. doi:10.1021/jo01350a537.
- Koide, S. G.; Barron, A. R. (1996). "Alumoxanes as Cocatalysts in the Palladium-Catalyzed Copolymerization of Carbon Monoxide and Ethylene: Genesis of a Structure-Activity Relationship". Organometallics. 15 (9): 2213. doi:10.1021/om9508492.
- Freixa, Z.; Van Leeuwen, P. W. N. M. (2003). "Bite angle effects in diphosphine metal catalysts: steric or electronic?". Dalton Trans. 2003 (10): 1890. doi:10.1039/b300322c.
- Kamer, P.; Van Leeuwen, P.; Reek, J. (2001). "Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis". Acc. Chem. Res. 34 (11): 895–904. doi:10.1021/ar000060. PMID 11714261.
- Casey, C. P.; Whiteker, G. T.; Melville, M. G.; Petrovich, L. M.; Gavney, J. A.; Powell, D. R. (1992). "Diphosphines with natural bite angles near 120° increase selectivity for n-aldehyde formation in rhodium-catalyzed hydroformylation". J. Am. Chem. Soc. 114 (2): 5535–5543. doi:10.1021/ja00040a008.
- Heck, R.; Breslow, D. (1961). "The Reaction of Cobalt Hydrotetracarbonyl with Olefins". J. Am. Chem. Soc. 83 (19): 4023. doi:10.1021/ja01480a017.
Further reading
- Klinger, R.; Chen, M.; Rathke, J.; Kramarz, K. (2007). "Effect of Phosphines on the Thermodynamics of the Cobalt-Catalyzed Hydroformylation System". Organometallics. 26 (2): 352. doi:10.1021/om060768d.