Beta cell
Beta cells (β cells) are a type of cell found in pancreatic islets that synthesize and secrete insulin and amylin. Beta cells make up 50–70% of the cells in human islets.[1] In patients with type I or type II diabetes, beta-cell mass and function are diminished, leading to insufficient insulin secretion and hyperglycemia.[2]
| Beta cell | |
|---|---|
| Details | |
| Location | Pancreatic islet |
| Function | Insulin secretion |
| Identifiers | |
| Latin | endocrinocytus B; insulinocytus |
| TH | H3.04.02.0.00026 |
| Anatomical terms of microanatomy | |
Function
The primary function of a beta cell is to produce and release insulin and amylin. Both are hormones which reduce blood glucose levels by different mechanisms. Beta cells can respond quickly to spikes in blood glucose concentrations by secreting some of their stored insulin and amylin while simultaneously producing more.[3]
Insulin synthesis
Beta cells are the only site of insulin synthesis in mammals.[4] As glucose stimulates insulin secretion, it simultaneously increases proinsulin biosynthesis, mainly through translational control.[3]
The insulin gene is first transcribed into mRNA and translated into preproinsulin.[3] After translation, the preproinsulin precursor contains an N-terminal signal peptide that allows translocation into the rough endoplasmic reticulum (RER).[5] Inside the RER, the signal peptide is cleaved to form proinsulin.[5] Then, folding of proinsulin occurs forming three disulfide bonds.[5] Subsequent to protein folding, proinsulin is transported to the Golgi apparatus and enters immature insulin granules where proinsulin is cleaved to form insulin and C-peptide.[5] After maturation, these secretory vesicles hold insulin, C-peptide, and amylin until calcium triggers exocytosis of the granule contents.[3]
Through translational processing, insulin is encoded as a 110 amino acid precursor but is secreted as a 51 amino acid protein.[5]
Insulin secretion
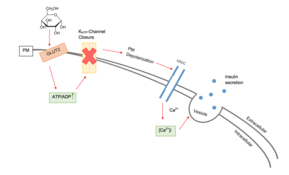
In beta cells, insulin release is stimulated primarily by glucose present in the blood.[3] As circulating glucose levels rise such as after ingesting a meal, insulin is secreted in a dose-dependent fashion.[3] This system of release is commonly referred to as glucose-stimulated insulin secretion (GSIS).[6] There are four key pieces to the "Consensus Model" of GSIS: GLUT2 dependent glucose uptake, glucose metabolism, KATP channel closure, and the opening of voltage gated calcium channels causing insulin granule fusion and exocytosis.[7]
Voltage-gated calcium channels and ATP-sensitive potassium ion channels are embedded in the plasma membrane of beta cells.[7][8] These ATP-sensitive potassium ion channels are normally open and the calcium ion channels are normally closed.[3] Potassium ions diffuse out of the cell, down their concentration gradient, making the inside of the cell more negative with respect to the outside (as potassium ions carry a positive charge).[3] At rest, this creates a potential difference across the cell surface membrane of -70mV.[9]
When the glucose concentration outside the cell is high, glucose molecules move into the cell by facilitated diffusion, down its concentration gradient through the GLUT2 transporter.[10] Since beta cells use glucokinase to catalyze the first step of glycolysis, metabolism only occurs around physiological blood glucose levels and above.[3] Metabolism of the glucose produces ATP, which increases the ATP to ADP ratio.[11]
The ATP-sensitive potassium ion channels close when this ratio rises.[8] This means that potassium ions can no longer diffuse out of the cell.[12] As a result, the potential difference across the membrane becomes more positive (as potassium ions accumulate inside the cell).[9] This change in potential difference opens the voltage-gated calcium channels, which allows calcium ions from outside the cell to diffuse in down their concentration gradient.[9] When the calcium ions enter the cell, they cause vesicles containing insulin to move to, and fuse with, the cell surface membrane, releasing insulin by exocytosis into the hepatic portal vein.[13][14]
Other hormones secreted
- C-peptide, which is secreted into the bloodstream in equimolar quantities to insulin. C-peptide helps to prevent neuropathy and other vascular deterioration related symptoms of diabetes mellitus.[15] A practitioner would measure the levels of C-peptide to obtain an estimate for the viable beta cell mass.[16]
- Amylin, also known as islet amyloid polypeptide (IAPP).[17] The function of amylin is to slow the rate of glucose entering the bloodstream. Amylin can be described as a synergistic partner to insulin, where insulin regulates long term food intake and amylin regulates short term food intake.
Clinical significance
Type 1 diabetes
Type 1 diabetes mellitus, also known as insulin dependent diabetes, is believed to be caused by an auto-immune mediated destruction of the insulin producing beta cells in the body.[5] The process of beta-cell destruction begins with insulitis activating antigen presenting cells (APCs). APCs then trigger activation of CD4+ helper-T cells, and chemokines/cytokines release. Then the cytokines activate CD8+ cytotoxic–T cells, which lead to beta-cell destruction.[18] The destruction of these cells reduces the body's ability to respond to glucose levels in the body, therefore making it nearly impossible to properly regulate glucose and glucagon levels in the bloodstream.[19] The body destroys 70–80% of beta cells, leaving only 20–30% of functioning cells.[2][20] This can cause the patient to experience hyperglycemia, which leads to other adverse short-term and long-term conditions.[21] The symptoms of diabetes can potentially be controlled with methods such as regular doses of insulin and sustaining a proper diet.[21] However, these methods can be tedious and cumbersome to continuously perform on a daily basis.[21]
Type 2 diabetes
Type 2 diabetes mellitus, also known as non insulin dependent diabetes and as chronic hyperglycemia, is caused primarily by genetics and the development of metabolic syndrome.[2][5] The beta cells can still secrete insulin but the body has developed a resistance and its response to insulin has declined.[3] It is believed to be due to the decline of specific receptors on the surface of the liver, adipose, and muscle cells which lose their ability to respond to insulin that circulates in the blood.[22][23] In an effort to secrete enough insulin to overcome the increasing insulin resistance, the beta cells increase their function, size and number.[3] Increased insulin secretion leads to hyperinsulinemia, but blood glucose levels remain within their normal range due to the decreased efficacy of insulin signaling.[3] However, the beta cells can become overworked and exhausted from being overstimulated, leading to a 50% reduction in function along with a 40% decrease in beta-cell volume.[5] At this point, not enough insulin can be produced and secreted to keep blood glucose levels within their normal range, causing overt type 2 diabetes.[5]
Insulinoma
Insulinoma is a rare tumor derived from the neoplasia of beta cells. Insulinomas are usually benign, but may be medically significant and even life-threatening due to recurrent and prolonged attacks of hypoglycemia.[24]
Medications
Many drugs to combat diabetes are aimed at modifying the function of the beta cell.
- Sulfonylureas are insulin secretagogues that act by closing the ATP-sensitive potassium channels, thereby causing insulin release.[25][26] These drugs are known to cause hypoglycemia and can lead to beta-cell failure due to overstimulation.[2] Second-generation versions of sulfonylureas are shorter acting and less likely to cause hypoglycemia.[26]
- GLP-1 receptor agonists stimulate insulin secretion by simulating activation of the body's endogenous incretin system.[26] The incretin system acts as an insulin secretion amplifying pathway.[26]
- DPP-4 inhibitors block DPP-4 activity which increases postprandial incretin hormone concentration, therefore increasing insulin secretion.[26]
Research
Experimental techniques
Many researchers around the world are investigating the pathogenesis of diabetes and beta-cell failure. Tools used to study beta-cell function are expanding rapidly with technology.
For instance, transcriptomics have allowed researchers to comprehensively analyze gene transcription in beta-cells to look for genes linked to diabetes.[2] A more common mechanism of analyzing cellular function is calcium imaging. Fluorescent dyes bind to calcium and allow in vitro imaging of calcium activity which correlates directly with insulin release.[2][27] A final tool used in beta-cell research are in vivo experiments. Diabetes mellitus can be experimentally induced in vivo for research purposes by streptozotocin[28] or alloxan,[29] which are specifically toxic to beta cells. Mouse and rat models of diabetes also exist including ob/ob and db/db mice which are a type 2 diabetes model, and non-obese diabetic mice (NOD) which are a model for type 1 diabetes.[30]
Type 1 diabetes
Research has shown that beta cells can be differentiated from human pancreas progenitor cells.[31] These differentiated beta cells, however, often lack much of the structure and markers that beta cells need to perform their necessary functions.[31] Examples of the anomalies that arise from beta cells differentiated from progenitor cells include a failure to react to environments with high glucose concentrations, an inability to produce necessary beta cell markers, and abnormal expression of glucagon along with insulin.[31]
In order to successfully re-create functional insulin producing beta cells, studies have shown that manipulating cell-signal pathways in early stem cell development will lead to those stem cells differentiating into viable beta cells.[31][32] Two key signal pathways have been shown to play a vital role in the differentiation of stem cells into beta cells: the BMP4 pathway and the kinase C.[32] Targeted manipulation of these two pathways has shown that it is possible to induce beta cell differentiation from stem cells.[32] These variations of artificial beta cells have shown greater levels of success in replicating the functionality of natural beta cells, although the replication has not been perfectly re-created yet.[32]
Studies have shown that it is possible to regenerate beta cells in vivo in some animal models.[33] Research in mice has shown that beta cells can often regenerate to the original quantity number after the beta cells have undergone some sort of stress test, such as the intentional destruction of the beta cells in the mice subject or once the auto-immune response has concluded.[31] While these studies have conclusive results in mice, beta cells in human subjects may not possess this same level of versatility. Investigation of beta cells following acute onset of Type 1 diabetes has shown little to no proliferation of newly synthesized beta cells, suggesting that human beta cells might not be as versatile as rat beta cells, but there is actually no comparison that can be made here because healthy (non-diabetic) rats were used to prove that beta cells can proliferate after intentional destruction of beta cells, while diseased (type-1 diabetic) humans were used in the study which was attempted to use as evidence against beta cells regenerating, which in reality tells us literally nothing whatsoever.
It appears that much work has to be done in the field of regenerating beta cells.[32] Just as in the discovery of creating insulin through the use of recombinant DNA, the ability to artificially create stem cells that would differentiate into beta cells would prove to be an invaluable resource to patients suffering from Type 1 diabetes. An unlimited amount of beta cells produced artificially could potentially provide therapy to many of the patients who are affected by Type 1 diabetes.
Type 2 diabetes
Research focused on non insulin dependent diabetes encompasses many areas of interest. Degeneration of the beta cell as diabetes progresses has been a broadly reviewed topic.[2][3][5] Another topic of interest for beta-cell physiologists is the mechanism of insulin pulsatility which has been well investigated.[34][35] Many genome studies have been completed and are advancing the knowledge of beta-cell function exponentially.[36][37] Indeed, the area of beta-cell research is very active yet many mysteries remain.
See also
- Gastric inhibitory polypeptide receptor
- List of terms associated with diabetes
- Guangxitoxin
- Alpha cell
- Pancreatic development
- Islets of Langerhans
References
- Dolenšek J, Rupnik MS, Stožer A (2015-01-02). "Structural similarities and differences between the human and the mouse pancreas". Islets. 7 (1): e1024405. doi:10.1080/19382014.2015.1024405. PMC 4589993. PMID 26030186.
- Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S (September 2017). "Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis". Molecular Metabolism. 6 (9): 943–957. doi:10.1016/j.molmet.2017.06.019. PMC 5605733. PMID 28951820.
- Boland BB, Rhodes CJ, Grimsby JS (September 2017). "The dynamic plasticity of insulin production in β-cells". Molecular Metabolism. 6 (9): 958–973. doi:10.1016/j.molmet.2017.04.010. PMC 5605729. PMID 28951821.
- Boland BB, Brown C, Alarcon C, Demozay D, Grimsby JS, Rhodes CJ (February 2018). "β-Cell Control of Insulin Production During Starvation-Refeeding in Male Rats". Endocrinology. 159 (2): 895–906. doi:10.1210/en.2017-03120. PMC 5776497. PMID 29244064.
- Fu Z, Gilbert ER, Liu D (January 2013). "Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes". Current Diabetes Reviews. 9 (1): 25–53. doi:10.2174/157339913804143225. PMC 3934755. PMID 22974359.
- Komatsu M, Takei M, Ishii H, Sato Y (November 2013). "Glucose-stimulated insulin secretion: A newer perspective". Journal of Diabetes Investigation. 4 (6): 511–6. doi:10.1111/jdi.12094. PMC 4020243. PMID 24843702.
- Ramadan JW, Steiner SR, O'Neill CM, Nunemaker CS (December 2011). "The central role of calcium in the effects of cytokines on beta-cell function: implications for type 1 and type 2 diabetes". Cell Calcium. 50 (6): 481–90. doi:10.1016/j.ceca.2011.08.005. PMC 3223281. PMID 21944825.
- Ashcroft FM, Rorsman P (February 1990). "ATP-sensitive K+ channels: a link between B-cell metabolism and insulin secretion". Biochemical Society Transactions. 18 (1): 109–11. doi:10.1042/bst0180109. PMID 2185070.
- MacDonald PE, Joseph JW, Rorsman P (December 2005). "Glucose-sensing mechanisms in pancreatic beta-cells". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 360 (1464): 2211–25. doi:10.1098/rstb.2005.1762. PMC 1569593. PMID 16321791.
- De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, Schuit F (November 1995). "Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression". The Journal of Clinical Investigation. 96 (5): 2489–95. doi:10.1172/JCI118308. PMC 185903. PMID 7593639.
- Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D'Ascia SL, Cannone M, Marziliano N, Trimarco B, Guise TA, Lacampagne A, Marks AR (May 2015). "Calcium release channel RyR2 regulates insulin release and glucose homeostasis". The Journal of Clinical Investigation. 125 (5): 1968–78. doi:10.1172/JCI79273. PMC 4463204. PMID 25844899.
- Keizer J, Magnus G (August 1989). "ATP-sensitive potassium channel and bursting in the pancreatic beta cell. A theoretical study". Biophysical Journal. 56 (2): 229–42. Bibcode:1989BpJ....56..229K. doi:10.1016/S0006-3495(89)82669-4. PMC 1280472. PMID 2673420.
- Lang V, Light PE (2010). "The molecular mechanisms and pharmacotherapy of ATP-sensitive potassium channel gene mutations underlying neonatal diabetes". Pharmacogenomics and Personalized Medicine. 3: 145–61. doi:10.2147/PGPM.S6969. PMC 3513215. PMID 23226049.
- Edgerton DS, Kraft G, Smith M, Farmer B, Williams PE, Coate KC, Printz RL, O'Brien RM, Cherrington AD (March 2017). "Insulin's direct hepatic effect explains the inhibition of glucose production caused by insulin secretion". JCI Insight. 2 (6): e91863. doi:10.1172/jci.insight.91863. PMC 5358484. PMID 28352665.
- Ido Y, Vindigni A, Chang K, Stramm L, Chance R, Heath WF, DiMarchi RD, Di Cera E, Williamson JR (July 1997). "Prevention of vascular and neural dysfunction in diabetic rats by C-peptide". Science. 277 (5325): 563–6. doi:10.1126/science.277.5325.563. PMID 9228006.
- Hoogwerf BJ, Goetz FC (January 1983). "Urinary C-peptide: a simple measure of integrated insulin production with emphasis on the effects of body size, diet, and corticosteroids". The Journal of Clinical Endocrinology and Metabolism. 56 (1): 60–7. doi:10.1210/jcem-56-1-60. PMID 6336620.
- Moore CX, Cooper GJ (August 1991). "Co-secretion of amylin and insulin from cultured islet beta-cells: modulation by nutrient secretagogues, islet hormones and hypoglycemic agents". Biochemical and Biophysical Research Communications. 179 (1): 1–9. doi:10.1016/0006-291X(91)91325-7. PMID 1679326.
- Tomita T. Apoptosis of pancreatic β-cells in Type 1 diabetes. Bosn J of Basic Med Sci. 2017;17(3):183-9. DOI:https://doi.org/10.17305/bjbms.2017.1961 PMCID: PMC5581966 PMID: 28368239
- Eizirik, D. L.; Mandrup-Poulsen, T. (2001-12-01). "A choice of death - the signal-transduction of immune-mediated beta-cell apoptosis". Diabetologia. 44 (12): 2115–2133. doi:10.1007/s001250100021. ISSN 0012-186X. PMID 11793013.
- Butler, A. E.; Galasso, R.; Meier, J. J.; Basu, R.; Rizza, R. A.; Butler, P. C. (2007-09-06). "Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis". Diabetologia. 50 (11): 2323–2331. doi:10.1007/s00125-007-0794-x. ISSN 0012-186X. PMID 17805509.
- Ciechanowski, Paul S.; Katon, Wayne J.; Russo, Joan E.; Hirsch, Irl B. (July–August 2003). "The relationship of depressive symptoms to symptom reporting, self-care and glucose control in diabetes". General Hospital Psychiatry. 25 (4): 246–252. doi:10.1016/s0163-8343(03)00055-0. ISSN 0163-8343. PMID 12850656.
- "U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group". Diabetes. 44 (11): 1249–58. November 1995. doi:10.2337/diabetes.44.11.1249. PMID 7589820.
- Rudenski AS, Matthews DR, Levy JC, Turner RC (September 1991). "Understanding "insulin resistance": both glucose resistance and insulin resistance are required to model human diabetes". Metabolism. 40 (9): 908–17. doi:10.1016/0026-0495(91)90065-5. PMID 1895955.
- Yu, Run; Nissen, Nicholas N.; Hendifar, Andrew; Tang, Laura; Song, Yu-Li; Chen, Yuan-Jia; Fan, Xuemo (January 2017). "A Clinicopathological Study of Malignant Insulinoma in a Contemporary Series". Pancreas. 46 (1): 48–56. doi:10.1097/MPA.0000000000000718. ISSN 1536-4828. PMID 27984486.
- Bolen, Shari; Feldman, Leonard; Vassy, Jason; Wilson, Lisa; Yeh, Hsin-Chieh; Marinopoulos, Spyridon; Wiley, Crystal; Selvin, Elizabeth; Wilson, Renee (2007-09-18). "Systematic Review: Comparative Effectiveness and Safety of Oral Medications for Type 2 Diabetes Mellitus". Annals of Internal Medicine. 147 (6): 386–99. doi:10.7326/0003-4819-147-6-200709180-00178. ISSN 0003-4819. PMID 17638715.
- Inzucchi, S. E.; Bergenstal, R. M.; Buse, J. B.; Diamant, M.; Ferrannini, E.; Nauck, M.; Peters, A. L.; Tsapas, A.; Wender, R. (2012-04-20). "Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD)". Diabetologia. 55 (6): 1577–1596. doi:10.1007/s00125-012-2534-0. ISSN 0012-186X. PMID 22526604.
- Whitticar, Nicholas B.; Strahler, Elisha W.; Rajan, Parthiban; Kaya, Savas; Nunemaker, Craig S. (2016-11-21). "An Automated Perifusion System for Modifying Cell Culture Conditions over Time". Biological Procedures Online. 18 (1): 19. doi:10.1186/s12575-016-0049-7. ISSN 1480-9222. PMC 5117600. PMID 27895534.
- Wang Z, Gleichmann H (January 1998). "GLUT2 in pancreatic islets: crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice". Diabetes. 47 (1): 50–6. doi:10.2337/diabetes.47.1.50. PMID 9421374.
- Danilova IG, Sarapultsev PA, Medvedeva SU, Gette IF, Bulavintceva TS, Sarapultsev AP (February 2015). "Morphological restructuring of myocardium during the early phase of experimental diabetes mellitus" (PDF). Anatomical Record. 298 (2): 396–407. doi:10.1002/ar.23052. hdl:10995/73117. PMID 25251897.
- King, Aileen JF (June 2012). "The use of animal models in diabetes research". British Journal of Pharmacology. 166 (3): 877–894. doi:10.1111/j.1476-5381.2012.01911.x. ISSN 0007-1188. PMC 3417415. PMID 22352879.
- Afelik, Solomon; Rovira, Meritxell (2017-04-15). "Pancreatic β-cell regeneration: Facultative or dedicated progenitors?". Molecular and Cellular Endocrinology. 445: 85–94. doi:10.1016/j.mce.2016.11.008. ISSN 1872-8057. PMID 27838399.
- Mahla RS (2016). "Stem Cells Applications in Regenerative Medicine and Disease Therapeutics". International Journal of Cell Biology. 2016 (7): 1–24. doi:10.1155/2016/6940283. PMC 4969512. PMID 27516776.
- Jeon, Kilsoo; Lim, Hyejin; Kim, Jung-Hyun; Thuan, Nguyen Van; Park, Seung Hwa; Lim, Yu-Mi; Choi, Hye-Yeon; Lee, Eung-Ryoung; Kim, Jin-Hoi (2012-09-20). "Differentiation and transplantation of functional pancreatic beta cells generated from induced pluripotent stem cells derived from a type 1 diabetes mouse model". Stem Cells and Development. 21 (14): 2642–2655. doi:10.1089/scd.2011.0665. ISSN 1557-8534. PMC 3438879. PMID 22512788.
- Nunemaker, Craig S.; Bertram, Richard; Sherman, Arthur; Tsaneva-Atanasova, Krasimira; Daniel, Camille R.; Satin, Leslie S. (2006-09-15). "Glucose modulates [Ca2+]i oscillations in pancreatic islets via ionic and glycolytic mechanisms". Biophysical Journal. 91 (6): 2082–2096. doi:10.1529/biophysj.106.087296. ISSN 0006-3495. PMC 1557567. PMID 16815907.
- Bertram, Richard; Sherman, Arthur; Satin, Leslie S. (2007-10-01). "Metabolic and electrical oscillations: partners in controlling pulsatile insulin secretion". American Journal of Physiology. Endocrinology and Metabolism. 293 (4): E890–E900. doi:10.1152/ajpendo.00359.2007. ISSN 0193-1849. PMID 17666486.
- Muraro, Mauro J.; Dharmadhikari, Gitanjali; Grün, Dominic; Groen, Nathalie; Dielen, Tim; Jansen, Erik; van Gurp, Leon; Engelse, Marten A.; Carlotti, Francoise (2016-10-26). "A Single-Cell Transcriptome Atlas of the Human Pancreas". Cell Systems. 3 (4): 385–394.e3. doi:10.1016/j.cels.2016.09.002. ISSN 2405-4712. PMC 5092539. PMID 27693023.
- Segerstolpe, Åsa; Palasantza, Athanasia; Eliasson, Pernilla; Andersson, Eva-Marie; Andréasson, Anne-Christine; Sun, Xiaoyan; Picelli, Simone; Sabirsh, Alan; Clausen, Maryam (2016-10-11). "Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes". Cell Metabolism. 24 (4): 593–607. doi:10.1016/j.cmet.2016.08.020. ISSN 1550-4131. PMC 5069352. PMID 27667667.
| Authority control |
|
|---|