Virial expansion
The classical virial expansion expresses the pressure of a many-particle system in equilibrium as a power series in the number density:


Here the quantity is the compressibility factor. This is the virial equation of state, the most general function relating PρT properties of fluids, first proposed by Kamerlingh Onnes.[1] The compressibility factor is a dimensionless quantity, indicating how much a real fluid deviates from an ideal gas. A is the first virial coefficient, which has a constant value of 1 and it makes the statement that at low molar density, all fluids behave like ideal gases. Virial coefficients B, C, D, etc., are temperature dependent, and are generally presented as Taylor series in terms of 1/T.

Second and third virial coefficients
The second and third virial coefficients had been studied extensively and tabulated for many fluids for more than a century. The most extensive compilation was in the books by Dymonds.[2][3] Recently, Thermodynamics Research Center of National Institute of Standards and Technology (NIST/TRC) published a huge amount of thermodynamics data in the forms of Web Thermo Tables (WTT).[4] In the WTT-Lite version, critically reviewed data on 150 fluids are available online. Tables of second and third virial coefficients of many fluids are also included in this compilation.
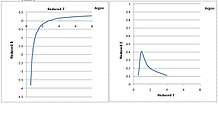
The second and third virial coefficients, as functions of temperature, of argon are shown in the following figure.[5] Reduced temperature and reduced virial coefficients, scaled by respective critical properties, are all dimensionless. Most fluids share the same behavior.
The second virial coefficient decreases monotonically as temperature is lowered. However, the third virial coefficient has a bell shape. It increases as temperature is lowered to the critical temperature, then it passes through a peak and decreases rapidly to 0 as temperature is lowered from the critical point to the triple point. It is physically unreasonable for it to decrease below the critical temperature, because the third virial coefficient theoretically represents the repulsive force among three molecules, which is expected to increase at lower temperature, as molecules are pressed together.
The reason why the third virial coefficient decreased below the critical temperature is that it had been analyzed incorrectly. Generally the PρT isotherms were measured conveniently in the gaseous phase. Below the critical temperature, gaseous phase condenses and coexists with liquid phase, and the PρT isotherm becomes flat. Saturation pressure does not change until gas condenses completely to liquid, and then pressure rises as density increases. There is a large gap between pure gaseous phase and pure liquid phase where no useful pressure data, except saturation pressure, are available. If only data in the gaseous phase were analyzed, the third virial coefficient became very small, because the PρT isotherm was almost linear in the gaseous phase. However, if data points in the pure liquid phase were included, a second order regression would give a large third virial coefficient. The third virial coefficient thus derived would increase monotonically as temperature is lowered from the critical point to the triple point.
The expectation that the third virial coefficient is a monotonically increasing function of 1/T can be verified with equations of state which accurately predicted the PρT isotherms in the saturation region where gaseous and liquid phases coexist. Most equations of state can be cast into a virial form, so that the second and third virial coefficients derived from them can be compare closely.
Casting equations of state into virial form
Most equations of state can be reformulated and cast in virial equations to evaluate and compare their implicit second and third virial coefficients. The seminal Van der Waals equation of state,[6] was proposed in 1873:

where v = 1/ρ is molar volume. It can be rearranged by expanding 1/(v - b) into a Taylor’s series:

The second virial coefficient has roughly the correct behavior, as it decreases monotonically when temperature is lowered. The third and higher virial coefficients are independent of temperature, and are definitely not correct, especially at low temperatures. Almost all subsequent equations of state derived from Van der Waals equation, like those from Dieterici,[7] Berthelot,[8] Redlich-Kwong,[9] Peng-Robinson,[10] etc., suffered from the singularity introduced by 1/(v - b), and could not be made to represent accurately the PρT isotherms at temperatures below critical temperature. Many of them produced adequate second virial coefficients, but most gave incorrect third virial coefficients.
The other school of equations of state, started by Beattie-Bridgeman,[11] however, were more closely related to virial equations, and shown to be more accurate in representing behavior of fluids in both gaseous and liquid phases. They can be easily reformulated into virial equations of state, and compared with one another. The Beattie-Bridgeman equation of state, proposed in 1928,

where


can be rearranged:

This equation of state represented the second virial coefficient very well. However, the third virial coefficient had the wrong sign. Hence it failed to represent isotherms close to and below the critical temperature.
The Benedict-Webb-Rubin equation of state[12] in 1940 was a significant improvement, in representing isotherms below the critical temperature:

More improvements were proposed by Starling[13] in 1972:

Following are plots of reduced second and third virial coefficients against reduced temperature according to Starling[13]:

The exponential terms in the last two equations seem intimidating, and out of line for a virial expansion sequence. Its purpose was to correct the third virial coefficient, so that the isotherms in the liquid phase could be represented correctly. Actually, the exponential term converges very rapidly as ρ increases, and if we took only the first two terms in its Taylor expansion series, , and multiply it with , the result is . It thus contributed a term to the third virial coefficient, and one term to the eighth virial coefficient, which can be effectively ignored.




After expansion of the exponential terms, the Benedict-Webb-Rubin and Starling equations of state have this interesting form:

The fourth and fifth virial coefficients are zero. After the third virial term, the next significant term is the sixth virial coefficient. It seems that the first three virial terms dominant the compressibility factor for fluids, down to , and up to .


In the original study in 1901 by Kamerlingh Onnes[1], he omitted the fourth virial coefficient D, and designated the higher terms as a residue in his virial equation. Unfortunately, the significance of the first three third virial terms has never been fully appreciated, and their effects on gaseous-liquid equilibrium were masked by other higher virial coefficients in the blind search of precision, with "multi-variable optimization" algorithms or the likes.
It is now clear why Benedict-Webb-Rubin improved on Beattie-Bridgeman equation of state by adding the complicated exponential term. They ought to have recognized that the third virial coefficient in the gaseous phase were small, but had to be large in the liquid phase. Instead of enlarging the third virial coefficient, they chose to add the strange-looking exponential term, whose sole purpose were to make the third virial coefficient larger at lower temperatures. The Taylor expansion of this exponential term reveals their true intentions.
Re-analyzing data reported by Starling[13], the virial coefficients are best represented:



b and c could be determined using simple second order regression analysis from experimental PρT isotherms. and could then be determined using third order regression analysis on b and c. could then be determined by analyzing residues in compressibility factor after the first three virial terms were removed from the virial equation. The data reported by Starling[13] are re-analyzed and the results are shown in the following table. These coefficients are all dimensionless since they are all scaled with critical molar volumes and critical temperature.



| Fluid | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methane | 0.440 | -1.171 | -0.236 | -0.210 | 0.364 | -0.275 | -0.014 | 0.396 | 0.0319 | 1.71E-03 |
| Ethane | 0.330 | -0.806 | -0.363 | -0.378 | 0.553 | -0.675 | -0.038 | 0.680 | 0.0461 | 2.63E-03 |
| Propane | 0.288 | -0.706 | -0.245 | -0.575 | 0.532 | -0.546 | -0.308 | 0.843 | 0.0334 | 1.89E-02 |
| n-butane | 0.377 | -0.916 | -0.115 | -0.610 | 0.547 | -0.519 | -0.347 | 0.871 | 0.0305 | 2.04E-02 |
| i-butane | 0.438 | -1.051 | -0.172 | -0.401 | 0.483 | -0.342 | -0.021 | 0.538 | 0.0194 | 1.19E-03 |
| n-pentane | 0.481 | -1.056 | -0.166 | -0.560 | 0.668 | -0.720 | -0.204 | 0.841 | 0.0411 | 1.17E-02 |
| i-pentane | 0.242 | -0.674 | -0.306 | -0.520 | 0.815 | -0.943 | -0.194 | 0.868 | 0.0484 | 9.99E-03 |
| n-heane | 0.435 | -0.636 | -0.358 | -0.759 | 0.848 | -1.275 | -0.105 | 1.120 | 0.0604 | 4.98E-03 |
| n-heptane | 0.493 | -0.798 | -0.636 | -0.428 | 0.589 | -0.738 | -0.017 | 0.814 | 0.0508 | 1.21E-03 |
| n-octane | 0.600 | -0.744 | -0.456 | -0.763 | 0.174 | -0.197 | -0.272 | 0.919 | 0.0144 | 1.99E-02 |
| Nitrogen | 0.502 | -1.380 | 0.092 | -0.333 | 0.400 | -0.276 | -0.027 | 0.322 | 0.0279 | 2.72E-03 |
| CO2 | 0.178 | -0.044 | -1.517 | 0.039 | 0.428 | -0.422 | -0.008 | 0.687 | 0.0490 | 9.52E-04 |
| H2S | 0.191 | -0.927 | -0.078 | -0.366 | 1.093 | -1.227 | -0.001 | 0.577 | 0.0578 | 8.37E-05 |










Cubic virial equation of state
It is very interesting that the three term virial equation or a cubic virial equation of state

has all the nicest attributes of Van der Waals equation of state, without its fatal singularity at v = b. Theoretically, the second virial coefficient represents bimolecular attraction forces, and the third virial term represents the repulsive forces among three molecules in close contact. Intuitively, we should expect B became negative at low temperature, while C would remain positive to counterbalance the effect of B and pushes Z and hence pressure to high values as ρ increases.
As mentioned before, this cubic virial equation of state has all the attributes of van der Waals equation of state, without the significant issue of a singularity at v = b. At the critical state, the coefficients B and C can be solved in close form. Imposing the critical conditions:
- and


the cubic virial equation can be solved to yield:
- , and :



is therefore 0.333, comparing to 0.375 from Van der Waals equation of state.

Between the critical point and the triple point is the saturation region of fluids. In this region, gaseous phase coexists with liquid phase under saturation pressure , and saturation temperature . Under saturation pressure, liquid phase has a molar volume of , and gaseous phase has a molar volume of . The corresponding molar densities are and . These are saturation properties needed to compute second and third virial coefficients.






A valid equation of state must produce an isotherm which crosses the horizontal line of at and , on . Under and , gas is in equilibrium with liquid. It means that the PρT isotherm must have three roots at . The cubic virial equation of state at is:

It can be rearranged as:

The factor is actually the volume of saturated gas according to the ideal gas law, and can be given a unique name :



In the saturation region, the cubic equation has three roots, and can be written alternatively as:

which can be expanded as:

is a volume of an unstable state between and . The cubic equations are identical. Therefore, from the linear terms in these equations, can be solved:



From the quadratic terms, B can be solved:

And from the cubic terms, C can be solved:

Since , and were tabulated for many fluids with as a parameter, it is a simple matter to compute B and C in the saturation region of these fluids. The results are generally in agreement with those computed from Benedict-Webb-Rubin and Starling equations of state. However, accuracy in B and C are critically dependent on the measurements of and , which are very difficult to measure accurately at low temperatures. The measurement errors thus introduced into B and C should be considered when one compares the values thus derived with those derived from second order regression analysis of PρT isotherms.
Gas-liquid-solid equilibrium
The cubic virial equation of state accurately represents the gas-liquid equilibrium of most substance from the critical point down to the triple point, where solid phase starts to appear. It is a simple matter to extend it to account for the gas-liquid- solid equilibrium:

In this virial equation, the first term represents the pressure generated by kinetic energy of molecules. The second term represents long range bimolecular attraction, and the third term represents short range tri-molecular repulsion. The second term pulls the PVT isotherm down as volume is reduced, while the third term pushes the isotherm up. When the temperature is below critical point, the PVT isotherm thus has an S shaped bent which allows a liquid phase to coexist with the prevalent gaseous phase.



Now, if we had a term to pull the PVT isotherm down in the liquid phase, and a terms to push it back up, a solid phase could be created, as these two terms producing another S shaped bend between liquid and solid. It was demonstrated [14] that such an S shaped bend could be synthesized using a -function like Lorentzian function over a van der Waals equation of state. Such an equation of state was ugly, and very difficult to manipulate mathematically. A virial equation shown above is much cleaner and easier to handle.



Argon is used to evaluate realistically this extended virial equation for gas-liquid-solid equilibrium. Data will be analyzed in the reduced forms. All PVT variables are scaled by their respective critical values. It is expect from the principle of corresponding states that the results would apply to other well behaved fluids. The relevant data of argon are summarized in the following table:
| Property | Value | Reduced value |
|---|---|---|
| Critical point volume (dm3/mol) | 0.07459 | 1 |
| Critical point temperature (K) | 150.687 | 1 |
| Critical point pressure (MPa) | 4.863 | 1 |
| Critical compressibility | 0.291 | 0.291 |
| Triple point vapor volume (dm3/mol) | 9.853 | 132.1 |
| Triple point liquid volume (dm3/mol) | 0.0282 | 0.378 |
| Triple point solid volume (dm3/mol) | 0.246 | 0.330 |
| Triple point temperature (K) | 83.8058 | 0.553 |
| Triple point pressure (MPa) | 0.06889 | 0.0142 |
When the variables P, V, and T are replaced by their reduced equivalents, , , and , the virial equation takes the following form:




where , , , , and . We will be concerned mostly with condition at the triple point of argon, where b = 3.424 and c = 1.152 from an earlier study.





must be slightly larger than the volume of solid argon, 0.33, and must be between the volumes of liquid and solid argon. Initially, is set to the volume of solid, to produce the last sharply rising edge of the isotherm where solid phase appears at very low volume. The exponential n must be then determined, so that the valley in the n-2n potential must fit between the volumes of solid (0.33) and liquid (0.378). After the exponential n is determined, the value of can be adjusted to satisfy the Gibbs Rule, which requires that the Gibbs free energy of liquid phase and that of solid phase must be equal under the triple point temperature and pressure.



To produce a solid phase in argon, the exponential value n must be very large, larger than 20; otherwise, the PVT isotherm would not bend to an S-shape between liquid and solid. The best estimation is that n = 30, , , and . The isotherm is shown in the right figure, in which three virial terms are plotted separately for clarity:







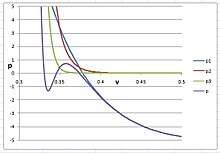
In this figure, represents the sum of the first three virial terms, of the cubic virial equation, and it shows the behavior of argon in its gaseous and liquid phases. represents the potential contributed from the term, and represents the contributions from the term. When n < 30, would interfere with and lower significantly the volume of liquid.





It was surprising that n has to be greater than 20 to produce a solid phase. It was well known that the Lenard-Jones 6-12 potential was used to compute the second virial coefficient from quantum mechanical principles. Quantum mechanical reasoning relates the second virial coefficient to bimolecular attraction, and the third coefficients to tri-molecular repulsion, etc. In the liquid phase of argon, one atom is surrounded by 12 nearest neighbors, and up to 32 next-to-nearest neighbors. In the solid phase, all atoms are locked in place, and the number of interacting neighbors is infinite. Therefore, n = 30, or even greater, is entirely reasonable, though surprising.
For the first time in the history of thermodynamics, we have a virial equation of state to describe quantitatively the gas-liquid-solid equilibrium for argon, and all fluids which observe the principle of corresponding states, at its triple point:

State of virial equations
With the advancement of computers, virial equations had been abused to represent large amounts of PρT data without understanding its coefficients. From Benedict-Webb-Rubin and Starling equations of state, it was determined that the best virial equation of state should take the form of :. The second and third virial coefficients in this equation can be computed from experimental PρT data using a simple linear regression available in Excel. After removing the first three virial terms, the residue in compressibility factor Z will then be analyzed to obtain the sixth virial coefficient.
References
- Kamerlingh Onnes H., Expression of state of gases and liquids by means of series, KNAW Proceedings, 4, 1901-1902, Amsterdam, 125-147 (1902).
- Dymond J. D., Wilhoit R. C., Virial coefficients of pure gases and mixtures, Springer (2003).
- Dymond J. H., Smith E. B., Virial coefficients of pure gases and mixtures. A critical compilation, Oxford University Press, 1st Edition (1969), 2nd Edition (1980).
- Lemmon, E.W., Huber, M.L., McLinden, M.O. NIST Standard Reference Database 23: Reference Fluid Thermodynamic and Transport Properties-REFPROP, Version 8.0, National Institute of Standards and Technology, Standard Reference Data Program: Gaithersburg, MD, (2007).
- Stewart R. B., Jacobsen R. T., Thermodynamic properties of argon from the triple point to 1200K with pressures to 1000 MPa, J. Phys. Chem. Ref. Data, Vol. 18, 639-798 (1989).
- Van der Waals J. D., On the continuity of the gaseous and liquid states (Doctoral dissertation). Universiteit Leiden (1873).
- Dieterici(7), C. Dieterici, Ann. Phys. Chem. Wiedemanns Ann. 69, 685 (1899).
- D. Berthelot, D., in Travaux et Mémoires du Bureau international des Poids et Mesures – Tome XIII (Paris: Gauthier-Villars, 1907).
- Redlich, Otto; Kwong, J. N. S. On The Thermodynamics of Solutions, Chem. Rev. 44 (1): 233–244 (1949).
- Peng, D. Y.; Robinson, D. B., A New Two-Constant Equation of State. Industrial and Engineering Chemistry: Fundamentals. 15: 59–64 (1976).
- Beattie, J. A., and Bridgeman, O. C., A new equation of state for fluids, Proc. Am. Acad. Art Sci., 63, 229-308 (1928).
- Benedict, Manson; Webb, George B.; Rubin, Louis C., An Empirical Equation for Thermodynamic Properties of Light Hydrocarbons and Their Mixtures: I. Methane, Ethane, Propane, and n-Butane, Journal of Chemical Physics, 8 (4): 334–345 (1940).
- Starling, Kenneth E., Fluid Properties for Light Petroleum Systems, Gulf Publishing Company, p. 270 (1973).
- Ting C. H., Chen C., Chen S., The gas-liquid-solid equilibrium studied by a simple equation of state, J. Chung Cheng Inst. Tech., Vol 3, No. 1, 77-84 (1972).