Thomas–Fermi screening
Thomas–Fermi screening is a theoretical approach to calculate the effects of electric field screening by electrons in a solid.[1] It is a special case of the more general Lindhard theory; in particular, Thomas–Fermi screening is the limit of the Lindhard formula when the wavevector (the reciprocal of the length-scale of interest) is much smaller than the fermi wavevector, i.e. the long-distance limit.[1] It is named after Llewellyn Thomas and Enrico Fermi.
The Thomas–Fermi wavevector (in Gaussian-cgs units) is[1]
- ,

where μ is the chemical potential (Fermi level), n is the electron concentration and e is the elementary charge.
Under many circumstances, including semiconductors that are not too heavily doped, n∝eμ/kBT, where kB is Boltzmann constant and T is temperature. In this case,
- ,

i.e. 1/k0 is given by the familiar formula for Debye length. In the opposite extreme, in the low-temperature limit T=0, electrons behave as quantum particles (fermions). Such an approximation is valid for metals at room temperature, and the Thomas–Fermi screening wavevector kTF given in atomic units is
- .

If we restore the electron mass and the Planck constant , the screening wavevector in Gaussian units is .



For more details and discussion, including the one-dimensional and two-dimensional cases, see the article on Lindhard theory.
Derivation
Relation between electron density and internal chemical potential
The internal chemical potential (closely related to Fermi level, see below) of a system of electrons describes how much energy is required to put an extra electron into the system, neglecting electrical potential energy. As the number of electrons in the system increases (with fixed temperature and volume), the internal chemical potential increases. This consequence is largely because electrons satisfy the Pauli exclusion principle: only one electron may occupy an energy level and lower-energy electron states are already full, so the new electrons must occupy higher and higher energy states.
The relationship is described by the electron number density as a function of μ, the internal chemical potential. The exact functional form depends on the system. For example, for a three-dimensional Fermi gas, a noninteracting electron gas, at absolute zero temperature, the relation is .


Proof: Including spin degeneracy,

(in this context—i.e., absolute zero—the internal chemical potential is more commonly called the Fermi energy).
As another example, for an n-type semiconductor at low to moderate electron concentration, .

Local approximation
The main assumption in the Thomas–Fermi model is that there is an internal chemical potential at each point r that depends only on the electron concentration at the same point r. This behaviour cannot be exactly true because of the Heisenberg uncertainty principle. No electron can exist at a single point; each is spread out into a wavepacket of size ≈ 1 / kF, where kF is the Fermi wavenumber, i.e. a typical wavenumber for the states at the Fermi surface. Therefore it cannot be possible to define a chemical potential at a single point, independent of the electron density at nearby points.
Nevertheless, the Thomas–Fermi model is likely to be a reasonably accurate approximation as long as the potential does not vary much over lengths comparable or smaller than 1 / kF. This length usually corresponds to a few atoms in metals.
Electrons in equilibrium, nonlinear equation
Finally, the Thomas–Fermi model assumes that the electrons are in equilibrium, meaning that the total chemical potential is the same at all points. (In electrochemistry terminology, "the electrochemical potential of electrons is the same at all points". In semiconductor physics terminology, "the Fermi level is flat".) This balance requires that the variations in internal chemical potential are matched by equal and opposite variations in the electric potential energy. This gives rise to the "basic equation of nonlinear Thomas–Fermi theory":[1]

where n(μ) is the function discussed above (electron density as a function of internal chemical potential), e is the elementary charge, r is the position, and is the induced charge at r. The electric potential is defined in such a way that at the points where the material is charge-neutral (the number of electrons is exactly equal to the number of ions), and similarly μ0 is defined as the internal chemical potential at the points where the material is charge-neutral.



Linearization, dielectric function
If the chemical potential does not vary too much, the above equation can be linearized:

where is evaluated at μ0 and treated as a constant.

This relation can be converted into a wavevector-dependent dielectric function:[1]

where

At long distances (q→0), the dielectric constant approaches infinity, reflecting the fact that charges get closer and closer to perfectly screened as you observe them from further away.
Example: A point charge
If a point charge Q is placed at r=0 in a solid, what field will it produce, taking electron screening into account?
One seeks a self-consistent solution to two equations:
- The Thomas–Fermi screening formula gives the charge density at each point r as a function of the potential at that point.
- The Poisson equation (derived from Gauss's law) relates the second derivative of the potential to the charge density.

For the nonlinear Thomas–Fermi formula, solving these simultaneously can be difficult, and usually there is no analytical solution. However, the linearized formula has a simple solution:

With k0=0 (no screening), this becomes the familiar Coulomb's law.
Note that there may be dielectric permittivity in addition to the screening discussed here; for example due to the polarization of immobile core electrons. In that case, replace Q by Q/ε, where ε is the relative permittivity due to these other contributions.
Fermi gas at arbitrary temperature
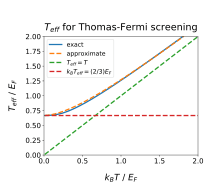
For a three-dimensional Fermi gas (noninteracting electron gas), the screening wavevector can be expressed as a function of both temperature and Fermi energy . The first step is calculating the internal chemical potential , which involves the inverse of a Fermi–Dirac integral,



- .

We can express in terms of an effective temperature : , or . The general result for is



.

In the classical limit , we find , while in the degenerate limit we find



- .

A simple approximate form that recovers both limits correctly is
- ,

for any power . A value that gives decent agreement with the exact result for all is [2], which has a maximum relative error of < 2.3%.



See also
References
- N. W. Ashcroft and N. D. Mermin, Solid State Physics (Thomson Learning, Toronto, 1976)
- Stanton, Liam G.; Murillo, Michael S. (2016-04-08). "Ionic transport in high-energy-density matter". Physical Review E. American Physical Society (APS). 93 (4): 043203. doi:10.1103/physreve.93.043203. ISSN 2470-0045.