Swain–Lupton equation
In physical organic chemistry, the Swain–Lupton equation is a linear free energy relationship (LFER) that is used in the study of reaction mechanisms and in the development of quantitative structure activity relationships for organic compounds. It was developed by C. Gardner Swain and Elmer C. Lupton Jr. in 1968 as a refinement of the Hammett equation to include both field effects and resonance effects.
Background
In organic chemistry, the Hammett plot provides a means to assess substituent effects on a reaction equilibrium or rate using the Hammett equation (1):
-
(1)

Hammett developed this equation from equilibrium constants from the dissociation of benzoic acid and derivatives (Fig. 1):
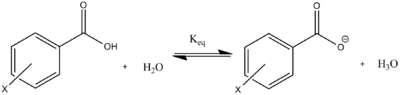
Hammett defined the equation based on two parameters: the reaction constant (ρ) and the substituent parameter (σ). When other reactions were studied using these parameters, a correlation was not always found due to the specific derivation of these parameters from the dissociation equilibrium of substituted benzoic acids and the original negligence of resonance effects. Therefore, the effects of substituents on an array of compounds must be studied on an individual reaction basis using the equation Hammett derived either for field or resonance effects, but not both.
Redefining the equation
C. Gardner Swain and Elmer C. Lupton Jr. from the Massachusetts Institute of Technology redefined the substituent parameter, σ, based on the idea that no more than two variables (resonance effects and field effects) are necessary to describe the effects of any given substituent. Field effects, F, are defined to include all effects (inductive and pure field). Likewise, effects due to resonance, R, are due to the average of electron-donating ability and electron-accepting ability. These two effects are assumed to be independent of each other and therefore can be written as a linear combination:
-
(2)

These two parameters are treated as independent terms because of the assumption that Swain and Lupton made; the substituent is kept distant by three or more saturated centers or if the substituent is (CH3)3N+. All other terms are then negligible and leads to the Swain–Lupton equation (2).
The new substituent parameter
The substituent parameter is now defined by field and resonance effects, F and R, which are dependent on the individual substituent. Constants r and f account for the importance of each of the two effects. These constants do not depend on the substituent but instead depend on the set of Hammett substituent parameters (σm, σp, σp+, σ', etc.).
In order to find the weighted constants, r and f, for each set of substituent parameters, one would need to establish the fact that each new substituent parameter σX could be written as a linear combination of specific reaction substituent parameters, i.e.
-
(3)

where σ1X and σ2X are specific substituent parameters (i.e. σ+, σ−, etc.) and c1 and c2 are constants independent of the substituent (depend on the reaction conditions, i.e. temperature, solvent, and individual reaction being studied). This can be expressed more generically as:
-
(4)

where i is an intercept to keep from fixing the origin at (0,0). If this was not done,the equation would give exceedingly more weight to the unsubstituted compounds that one is trying to make a comparison to using this equation.[1] A linear least-squares analysis is used to determine the coefficients/constants a, b, and i (Swain and Lupton used a procedure called DOVE: Dual Obligate Vector Evaluation).[2] Constants were first based on three previous reactions (σm, σp, σp+), which leads to more possible errors since the compiled data is only a minimal combination of a much larger pool. Seeing possible error in this limited pool, the data pool was increased by assigning a scale to begin with. A zero-scale is used for hydrogen, because it is known to neither readily donate or accept electron density when attached to a carbon atom due to similar electronegativities. A value of 1 was assigned to NO2, because previous research determined the effect of this substituent was predominantly due to resonance.[3] Lastly, F was set equal to R for both components so that the field effects could be compared directly to the resonance effects. This then leads to:
- F = R = 0 for H (Hydrogen).
- F = R = 1 for NO2 (Nitrogen Dioxide).
Fig. 2 shows some relative F and R values that Swain and Lupton founded.[2]
Substituent categories
Alkyl groups have a low to zero value for F but sensible values for R. This is most commonly explained by hyperconjugation, meaning little to no inductive effects but partial resonance effects.
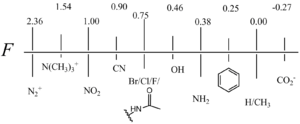
CF3 has a much higher R/F ratio than other substituents with high degrees of conjugation. This was studied in greater detail by Swain but is still explained best by fluoride hyperconjugation.
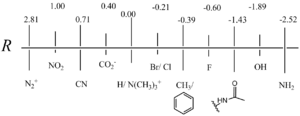
Positively charged substituents (i.e. N(CH3)3+ and S(CH3)2+) have larger positive F values due to a positive charge that is saturated near the carbon framework in question. Negatively charged substituents (i.e. CO2− and SO3−) have much lower F values because of their ability to resonate electron density amongst the oxygen atoms and stabilize it through hydrogen-bonding with solvents.
Linear free energy relationships are still useful, despite their disadvantages when pushed to the limits. New techniques to solve for Swain–Lupton substituent parameters involve studying chemical shifts through nuclear magnetic resonance spectroscopy. Recently, 15N NMR chemical shifts and substituent effects of 1,2,3,4,5,6,7,8-octahydroacridine and derivatives were studied. Values for R and F were found for the –N(COCH3)2 group, which could not be found previously using known methods.[4]
Values of f and r
It is sometime useful to look at the percent resonance (%r), because r is dependent on the reaction and is the same for all substituents.
-
(5)

One can predict the difference in data comparing two substituents using %r:
-
(6)

The most dominant effect is clear when looking at the ratio of R to F. For example, a tungsten complex was shown to alkylate allyl carbonates A and B. The ratio of products A1 and B1 can be attributed to the para substituent, X (Fig. 3). Using Swain–Lupton parameters (σ = 0.2F + 0.8R) a ρ value of -2.5 was found to be the slope.
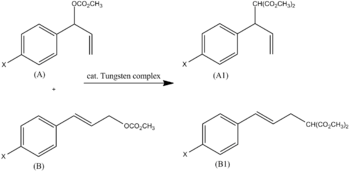
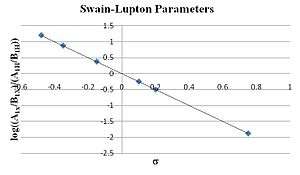
This is in agreement with the proposed mechanism (a positive charge forms on the benzylic carbon and is stabilized by resonance; R dominates by a ratio of 0.8/0.2).[5]
Disadvantages
Like any other linear free-energy relationship established, the Swain–Lupton equation will too fail when special circumstances arise, i.e. change in the rate determining step of a mechanism or solvation structure.[6]
See also
References
- Swain, C.G; Lupton, E.C., Jr. (1968). "Field and Resonance Components of Substituent Effects". J. Am. Chem. Soc. 90: 4328–4337. doi:10.1021/ja01018a024.CS1 maint: multiple names: authors list (link)
- Swain, C.G; Unger, S.H.; Rosenquist, N.R.; Swain, M.S. (1983). "Substituent Effects on Chemical Reactivity. Improved Evaluation of Field and Resonance Components". J. Am. Chem. Soc. 105: 492–502. doi:10.1021/ja00341a032.
- Wheland, G.W. (1955). Resonance in Organic Chemistry. New York: Wiley. pp. 367–368. ASIN B00005XST0.
- Potmischil,F.; Marinescu,M.; Nicolescu, A.; Deleanu, C.; Hillebrand, M (2008). "Hydroacridines: part 29. 15N NMR chemical shifts of 9-substituted 1,2,3,4,5,6,7,8-octahydroacridines and their N-oxides - Taft, Swain-Lupton, and other types of linear correlations". Magn. Reson. Chem. 46: 1141–1147. doi:10.1002/mrc.2335.
- Lehman, J.; Lloyd-Jone, G.C. (1995). "Regiocontrol and Stereoselectivity in Tungsten-Bipyridine Catalyzed Allylic Alkylation". Tetrahedron. 51: 8863–8874. doi:10.1016/0040-4020(95)00481-M.
- Swain, C.G. (1984). "Substituent and Solvent Effects on Chemical Reactivity". J. Org. Chem. 49: 2005–2010. doi:10.1021/jo00185a035.