Spike-and-wave
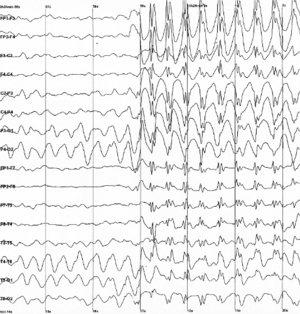
Spike-and-wave is a pattern of the electroencephalogram (EEG) typically observed during epileptic seizures. A spike-and-wave discharge is a regular, symmetrical, generalized EEG pattern seen particularly during absence epilepsy, also known as ‘petit mal’ epilepsy.[1] The basic mechanisms underlying these patterns are complex and involve part of the cerebral cortex, the thalamocortical network, and intrinsic neuronal mechanisms.[2] The first spike-and-wave pattern was recorded in the early twentieth century by Hans Berger. Many aspects of the pattern are still being researched and discovered, and still many aspects are uncertain. The spike-and-wave pattern is most commonly researched in absence epilepsy, but is common in several epilepsies such as Lennox-Gastaut syndrome (LGS) and Ohtahara syndrome. Antiepileptic drugs (AEDs) are commonly prescribed to treat epileptic seizures, and new ones are being discovered with fewer adverse effects. Today, most of the research is focused on the origin of the generalized bilateral spike-and-wave discharge. One proposal suggests that a thalamocortical (TC) loop is involved in the initiation spike-and-wave oscillations. Although there are several theories, the use of animal models has provided new insight on spike-and-wave discharge in humans.[3]
History
History of generalized epilepsy with absence seizures are dated to the eighteenth century, however the inventor of the electroencephalogram (EEG), Hans Berger, recorded the first EEG of an absence seizure in the 1920s, which led the way for the general notion of spike-and-wave electrophysiology. His first recording of a human EEG was made in 1924 using a galvanometer, but his results were very crude and showed small, undefined oscillations. He continued to refine his technique and increase the sensitivity to the galvanometer, in which he accumulated many EEGs of individuals with and without a brain malfunction or disorder. Among those tested were patients with epilepsy, dementia, and brain tumors.[4] Hans Berger published his findings in 1933, however his results did not give a definitive characterization of the general EEG pattern seen during an epileptic seizure. In 1935, F.A. Gibbs, H. Davis, and W.G. Lennox provided a clear description of EEG spike-and-wave patterns during a petit mal epileptic seizure.[5] An intracellular recording performed by DA Pollen in 1964 revealed that the "spike" aspect of the phenomenon was associated with neuronal firing and the "wave" aspect was associated with hyperpolarization.[6]
Pathophysiology
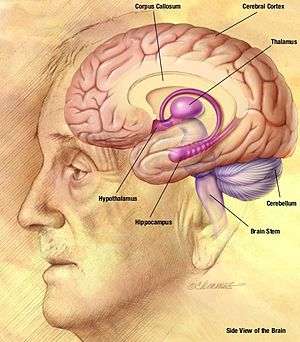
The spike-and-wave pattern seen during an absence seizure is the result of a bilateral synchronous firing of neurons ranging from the neocortex (part of the cerebral cortex) to the thalamus, along the thalamocortical network.[2] The EEG “spike” of the spike-and-wave complex corresponds to the depolarization of the neuronal membrane potential, also called a paroxysmal depolarizing shift (PDS). The initial understanding behind the mechanism of the PDS was that it was caused by a very large EPSP (excitatory postsynaptic potential) in the absence of synaptic inhibition, which relayed the action potentials in the neurons by triggering activation of voltage-gated channels. The voltage-gated sodium channels cause transitory sodium current into the cell, which generates the action potential. The voltage-gated calcium channels also have some effect on the depolarization of the cell, but the effect is minimal compared to the sodium channels. However, the increasing concentration of intracellular calcium leads to greater activation of calcium-activated potassium channels. These calcium-activated potassium channels, along with the voltage-gated potassium channels, contribute to the repolarization and hyperpolarization of the membrane. In an epileptic seizure, there are periods of a sustained depolarization, which cause a train of action potentials followed by a repolarization and hyperpolarization phase. The train of action potentials constitutes the “spike” phase, and the repolarization and hyperpolarization constitute the “wave” phase.[7]
Although there is evidence for the generation of a large EPSP, many studies have shown that synaptic inhibition remains functional during the generation of these types of paroxysmal depolarizing shifts.[8][9] Also, it has been shown that a decrease in the inhibitory activity does not affect neocortical kindling.[10] Therefore, the theory that spike-and-wave activity is caused by a giant EPSP due to the decrease or the absence of IPSPs (inhibitory postsynaptic potentials) is not accepted as a general mechanism for epileptic activity. Many studies have shown that the inhibitory postsynaptic signaling is actually increased during these epileptic attacks.[9] The activation of postsynaptic GABAA receptors leads to an increase in the intracellular chloride concentration, which in non-epileptic situations would lead to an IPSP. However, in seizure-related depolarizing shifts, there is a substantial activation of postsynaptic GABAA receptors, which leads to an even larger concentration of intracellular chloride concentration. This change in ion concentration gradient causes the GABAA inhibitory current to surpass the reversal potential, leading to an efflux of the chloride ions. This leads to a decreased amplitude or even reversed polarity of the IPSPs.[7]
Metabotropic glutamate receptors (mGluRs) in the thalamocortical network have also shown to display some role in the generation of spike-and-wave discharges (SWDs) associated with absence epilepsy. The different subtypes of mGlu receptors have a modulatory role on either excitatory or inhibitory synaptic transmission. There are conflicting hypotheses for the function of the many mGlu receptors with regards to epileptic seizures, however the role of the mGlu4 receptor is undisputed in the generation of SWDs, shown in animal models.[11] In one study, knockout mice lacking mGlu4 receptors showed a disruption of glutamate and GABA release in the thalamocortical network and were resistant to absence seizures induced by low doses of pentylenetetrazole.[12] Another study showed that bilateral injection of a mGlu4 receptor antagonist into the nRT (thalamic reticular nucleus) of normal mice protected against pentylenetetrazole induced seizures.[12] Also, WAG/Rij rats show an increased expression of mGlu4 receptors in the nRT when compared to a control group of normal rats.[13] These studies show that an increase in the expression and/or activity of mGlu4 receptors is associated with spike-and-wave discharges seen in absence seizures. This link between mGlur4 receptors and SWDs has led to the search for a selective mGlu4 receptor antagonist (which will block these receptors) as a potential new drug for the treatment of absence epilepsy.[11]
Initiation factors
The use of animal models, such as cats, for studying spike-and-wave discharges, has provided useful data for studying epilepsy in humans. One method of inducing a seizure in a cat is to inject penicillin into the cortical region of the brain. The spike-and-wave discharges seen in feline generalized penicillin epilepsy (FGPE) are very similar to the spike-and-wave discharges of a human absence seizure.[14] The use of rats has also been a common method for studying the spike-and-wave phenomenon. The Genetic Absence Epilepsy Rats from Strasbourg (GAERS) and the inbred Wistar Albino Glaxo rats from Rijswijk (WAG/Rij) are the two main strains of rats that have been used in studies. The rats from these two strains show spontaneously occurring absence seizures that consist of typical spike-and-wave activity seen on an EEG.[1] Rat genetic models have given data showing that the expression of absence seizures involves both the thalamic and cortical networks. In both models, electrophysiological data showed that spike-and-waves are initiated in the somatosensory cortex and then spread rapidly to the motor cortex and thalamic nuclei.[15][16] Using in vivo intracellular recordings, it was found in the GAERS that spike-and-wave are initiated in layer 5/6 neurons of the somatosensory cortex. These neurons, show a distinctive hyperactivity associated with a membrane depolarization. They are suggested to lead the firing of distant cortical cells during the epileptic discharge.[16]
Another possible initiation pattern tested in rats suggested the thalamocortical (TC) loop is involved in the initiation of spike-and-wave oscillations under certain conditions. In this study, relay and reticular thalamic neurons of epileptic and non-epileptic rats were dual extracellularly recorded and juxtacellularly labeled.[3] Medium oscillations (5–9 Hz) in both types of rats were noted to occur randomly in an unsynchronized pattern in relay and reticular neurons. However, spontaneous spike-and-wave discharges were observed in epileptic rats when the medium oscillations became synchronized, suggesting a dependence of the two. However, since medium ranged oscillations only developed into spike-and-wave discharges spontaneously, genetic factors also seem to contribute to the initiation of synchronized oscillations. These genetic factors may contribute to spike-and-wave oscillations by decreasing the action potential threshold in reticular cells, making them more excitable and potentially easier to initiate synchronized firing.[3] Another study has shown that these medium oscillations have led to spike-and-wave discharges.[17] The activity of the primary and secondary cortical regions, as well as the adjacent insular cortex were recorded using an EEG and where applied with electrical stimulation. The findings here showed that the onset of spike-and-wave discharged were followed by 5–9 Hz oscillations in these cortical regions as well.[17]
Genetic/developmental factors
Elongator Protein Complex 4 (ELP4) has been identified as a key component in the transcription of genes known to regulate the actin cytoskeleton, cell motility and migration of neurons. Research on ELP4 has been linked the gene to a centrotemporal sharp spike phenotype. Hypotheses have been made that a mutation in the non-coding region of the ELP4 gene may interfere with elongo-mediated gene interaction, specifically during the developmental stages of the cortical region.[18] This mutation may be responsible for a predisposition to spike-and-wave discharges, as well as other neurodevelopmental disorders.
Another study revealed that glucose may also be relevant to spike-and-wave occurrence in mice that contained a knock-in of the human GABA(A) γ2(R43Q) mutation, which has been known to be a genetic factor involved in the causation of absence epilepsy.[19] These absence seizure prone mice were injected with insulin to lower blood glucose levels by 40%. This reduction in blood glucose led to double the occurrence of spike-and-wave activity. Similar to the insulin effect, overnight fasting, where blood glucose levels were reduced by 35% also showed this double in occurrence. This model concludes that low glucose levels could be a potential trigger for absence seizures, and could be an environmental risk factor for humans.[19]
Spike-and-wave in epilepsy
Absence epilepsy
Bursts of generalized spike-and-wave discharges lasting two seconds or longer is considered an absence seizure.[20] Absence seizures are generalized epileptic seizures that can be divided into two types, typical and atypical. Typical and atypical absence seizures display two different kinds of spike-and-wave patterns. Typical absence seizures are described by generalized spike-and-wave patterns on an EEG with a discharge of 2.5 Hz or greater. They can be characterized by an increase in synchronization of discharges in the thalamocortical circuitry. They can also be characterized by the acute onset and termination of the seizure. Atypical absence seizures have a higher frequency in children with severe epilepsy that suffer from multiple types of seizures. The spike-and-wave pattern seen here is more irregular than the generalized pattern and also seems to be slower. This irregular pattern is due to non-synchronous discharges of the thalamocortical circuitry. The onset and termination in these atypical absence seizures seem to be less acute than the typical absence seizures.[21]
Lennox-Gastaut syndrome
Epileptic encephalopathies are a group of conditions that result in deterioration of sensory, cognitive, and motor functions due to consistent epileptic activity. Lennox-Gastaut syndrome (LGS) is a childhood epileptic encephalopathy characterized with generalized seizures and slow spike-wave activity while awake. LGS is a combination of atonic absences, tonic seizures, cognitive deterioration, and slow spike-wave activity in the EEG. This syndrome usually results from focal, multifocal, or diffuse brain damage and can be divided into symptomatic and cryptogenic types. Cognitive deterioration with high-frequency spike-wave activity affects most patients 2–9 years old with generalized seizures. The age of onset for LGS is between 1 and 10 years, between 2 and 6 years for symptomatic cases and 5 and 8 years for cryptogenic cases. Episodes can be triggered by modifications of treatment, which usually involves benzodiazepines, or changes in the conditions of life.[22]
Ohtahara syndrome
Ohtahara syndrome (OS), also known as early infantile epileptic encephalopathy (EIEE) with suppression-burst (S-B), is the most severe and the earliest-developing epileptic encephalopathy in children. This syndrome is characterized on an EEG by high voltage bursts and slow waves mixed with multifocal spikes alternating with almost flat suppression phases. The S-B will gradually begin to taper away at 3 months and disappear by 6 months. OS will transition to West syndrome or LGS with age. Tonic spasms are the main seizures observed in OS. Unlike LGS, the spike-and-wave pattern is consistent during both waking and sleeping states.[23] Symptoms of OS include:[24]
- Genetic defects
- Mitochondrial disease
- Mitochondrial respiratory chain defects
- Inborn errors of metabolism
- Glycine encephalopathy
- Cortical malformations
- Cerebral asymmetry
- Posterior fossa anomalies
- Agenesis of mammillary bodies
- Frequent minor generalized seizures
- Severe and continuous epileptic EEG abnormality
- Severe psychomotor prognosis
Spike-and-wave pattern during sleep
In continuous spike-and-wave syndrome (CSWS), a rare form of age-related epilepsy, children between the ages of three and seven exhibit continuous spike-and-wave discharges during slow-sleep. This disorder is found in 0.2–0.5% of all child epilepsy cases. This disorder's discharges rarely result in absence seizures, but motor impairment and neurophysiological regression have been found in CSWS. Spike-and-wave activity occupies about 85% of the non-rapid eye movement sleep.[25] This continuous pattern during sleep, like other aspects of spike-and-wave activity, are not completely understood either. However, what is hypothesized is that corticothalamic neuronal network that is involved in oscillating sleep patterns may begin to function as a pathologic discharging source.[18]
Clinical relevance
Reoccurrence after a solitary unprovoked seizure in children is about 50%, so the use of anti-epileptic drugs (AEDs) is very prevalent. AEDs aim to slow down the excess firing, associated with spike-and-wave discharges, at the beginning of seizures. They can bring about serious adverse drug reactions so physicians need to be aware of the safety and admissibility for each drug. These adverse effects are a major source of disability, morbidity, and mortality. Some of the adverse effects, such as serious cutaneous, haematological and hepatic events, usually require withdrawal in children and place a heavy burden on the costs of healthcare.[26]
Bromide was introduced as the first anti-epileptic drug 150 years ago. Because of the adverse effects mentioned above, bromide is not currently in use as an AED. Early treatment discontinuation was occurring far too frequently and eventually resulted in negative effects on several patients. Current treatment options include phenytoin, valproic acid, ethosuximide, and the new anti-epileptic drugs. Over the past 20 years, 15 new anti-epileptic drugs with positive outcomes have been introduced to the public. These new AEDs are aimed at improving the cost-benefit balance in AED therapy, improving tolerability profiles and reducing potential for drug interaction.[27] Despite these major advances, there is always room for improvement, especially regarding the tailored treatment of individuals who have suffered adverse effects from older AEDs.[26][28]
References
- Akman, Ozlem; Demiralp, Tamer; Ates, Nurbay; Onat, Filiz Yilmaz (2010). "Electroencephalographic differences between WAG/Rij and GAERS rat models of absence epilepsy". Epilepsy Research. 89 (2–3): 185–93. doi:10.1016/j.eplepsyres.2009.12.005. PMID 20092980.
- Snead, O. Carter (1995). "Basic mechanisms of generalized absence seizures". Annals of Neurology. 37 (2): 146–57. doi:10.1002/ana.410370204. PMID 7847856.
- Pinault, D; Vergnes, M; Marescaux, C (2001). "Medium-voltage 5–9-Hz oscillations give rise to spike-and-wave discharges in a genetic model of absence epilepsy: In vivo dual extracellular recording of thalamic relay and reticular neurons". Neuroscience. 105 (1): 181–201. doi:10.1016/S0306-4522(01)00182-8. PMID 11483311.
- Millett, David (2001). "Hans Berger: From Psychic Energy to the EEG". Perspectives in Biology and Medicine. 44 (4): 522–42. doi:10.1353/pbm.2001.0070. PMID 11600799.
- Avoli, Massimo (2012). "A brief history on the oscillating roles of thalamus and cortex in absence seizures". Epilepsia. 53 (5): 779–89. doi:10.1111/j.1528-1167.2012.03421.x. PMC 4878899. PMID 22360294.
- Pollen, D. A (1964). "Intracellular Studies of Cortical Neurons During Thalamic Induced Wave and Spike". Electroencephalography and Clinical Neurophysiology. 17 (4): 398–404. doi:10.1016/0013-4694(64)90163-4. PMID 14236822.
- Bazhenov, Maxim; Timofeev, Igor; Fröhlich, Flavio; Sejnowski, Terrence J (2008). "Cellular and network mechanisms of electrographic seizures". Drug Discovery Today: Disease Models. 5 (1): 45–57. doi:10.1016/j.ddmod.2008.07.005. PMC 2633479. PMID 19190736.
- Cohen, I; Navarro, V; Clemenceau, S; Baulac, M; Miles, R (2002). "On the Origin of Interictal Activity in Human Temporal Lobe Epilepsy in Vitro". Science. 298 (5597): 1418–21. doi:10.1126/science.1076510. PMID 12434059.
- Timofeev, I; Grenier, F; Steriade, M (2002). "The role of chloride-dependent inhibition and the activity of fast-spiking neurons during cortical spike–wave electrographic seizures". Neuroscience. 114 (4): 1115–32. doi:10.1016/S0306-4522(02)00300-7. PMID 12379264.
- Denslow, Maria J; Eid, Tore; Du, Fu; Schwarcz, Robert; Lothman, Eric W; Steward, Oswald (2001). "Disruption of Inhibition in Area CA1 of the Hippocampus in a Rat Model of Temporal Lobe Epilepsy". Journal of Neurophysiology. 86 (5): 2231–45. doi:10.1152/jn.2001.86.5.2231. PMID 11698514.
- Ngomba, Richard Teke; Santolini, Ines; Salt, Thomas E; Ferraguti, Francesco; Battaglia, Giuseppe; Nicoletti, Ferdinando; Van Luijtelaar, Gilles (2011). "Metabotropic glutamate receptors in the thalamocortical network: Strategic targets for the treatment of absence epilepsy". Epilepsia. 52 (7): 1211–22. doi:10.1111/j.1528-1167.2011.03082.x. PMID 21569017.
- Snead, O. Carter; Banerjee, P. K; Burnham, Mcintyre; Hampson, David (2000). "Modulation of Absence Seizures by the GABAAReceptor: A Critical Role for Metabotropic Glutamate Receptor 4 (mGluR4)". The Journal of Neuroscience. 20 (16): 6218–24. doi:10.1523/JNEUROSCI.20-16-06218.2000. PMID 10934271.
- Ngomba, R.T; Ferraguti, F; Badura, A; Citraro, R; Santolini, I; Battaglia, G; Bruno, V; De Sarro, G; Simonyi, A; Van Luijtelaar, G; Nicoletti, F (2008). "Positive allosteric modulation of metabotropic glutamate 4 (mGlu4) receptors enhances spontaneous and evoked absence seizures". Neuropharmacology. 54 (2): 344–54. doi:10.1016/j.neuropharm.2007.10.004. PMID 18022649.
- Giaretta, D; Avoli, M; Gloor, P (1987). "Intracellular recordings in pericruciate neurons during spike and wave discharges of feline generalized penicillin epilepsy". Brain Research. 405 (1): 68–79. doi:10.1016/0006-8993(87)90990-5. PMID 3032351.
- Meeren, Hanneke K. M; Pijn, Jan Pieter M; Van Luijtelaar, Egidius L. J. M; Coenen, Anton M. L; Lopes Da Silva, Fernando H (2002). "Cortical Focus Drives Widespread Corticothalamic Networks during Spontaneous Absence Seizures in Rats". The Journal of Neuroscience. 22 (4): 1480–95. doi:10.1523/JNEUROSCI.22-04-01480.2002. PMID 11850474.
- Polack, P.-O; Guillemain, I; Hu, E; Deransart, C; Depaulis, A; Charpier, S (2007). "Deep Layer Somatosensory Cortical Neurons Initiate Spike-and-Wave Discharges in a Genetic Model of Absence Seizures". Journal of Neuroscience. 27 (24): 6590–9. doi:10.1523/JNEUROSCI.0753-07.2007. PMID 17567820.
- Zheng, Thomas W; o'Brien, Terence J; Morris, Margaret J; Reid, Christopher A; Jovanovska, Valentina; o'Brien, Patrick; Van Raay, Leena; Gandrathi, Arun K; Pinault, Didier (2012). "Rhythmic neuronal activity in S2 somatosensory and insular cortices contribute to the initiation of absence-related spike-and-wave discharges". Epilepsia. 53 (11): 1948–58. doi:10.1111/j.1528-1167.2012.03720.x. PMID 23083325.
- Loddenkemper, Tobias; Fernández, Iván Sánchez; Peters, Jurriaan M (2011). "Continuous Spike and Waves During Sleep and Electrical Status Epilepticus in Sleep". Journal of Clinical Neurophysiology. 28 (2): 154–64. doi:10.1097/WNP.0b013e31821213eb. PMID 21399511.
- Reid, Christopher A; Kim, Tae Hwan; Berkovic, Samuel F; Petrou, Steven (2011). "Low blood glucose precipitates spike-and-wave activity in genetically predisposed animals". Epilepsia. 52 (1): 115–20. doi:10.1111/j.1528-1167.2010.02911.x. PMID 21175610.
- Szaflarski, Jerzy P; Difrancesco, Mark; Hirschauer, Thomas; Banks, Christi; Privitera, Michael D; Gotman, Jean; Holland, Scott K (2010). "Cortical and subcortical contributions to absence seizure onset examined with EEG/fMRI". Epilepsy & Behavior. 18 (4): 404–13. doi:10.1016/j.yebeh.2010.05.009. PMC 2922486. PMID 20580319.
- Velazquez, Jose L. Perez; Huo, Jeanne Zhen; Dominguez, L. Garcia; Leshchenko, Yevgen; Snead Iii, O. Carter (2007). "Typical versus Atypical Absence Seizures: Network Mechanisms of the Spread of Paroxysms". Epilepsia. 48 (8): 1585–93. doi:10.1111/j.1528-1167.2007.01120.x. PMID 17484751.
- Dulac, Olivier (2001). "Epileptic Encephalopathy". Epilepsia. 42: 23–6. doi:10.1046/j.1528-1157.2001.042suppl.3023.x. PMID 11520318.
- Ohtahara, Shunsuke; Yamatogi, Yasuko (2006). "Ohtahara syndrome: With special reference to its developmental aspects for differentiating from early myoclonic encephalopathy". Epilepsy Research. 70: S58–67. doi:10.1016/j.eplepsyres.2005.11.021. PMID 16829045.
- Pavone, Piero; Spalice, Alberto; Polizzi, Agata; Parisi, Pasquale; Ruggieri, Martino (2012). "Ohtahara syndrome with emphasis on recent genetic discovery". Brain and Development. 34 (6): 459–68. doi:10.1016/j.braindev.2011.09.004. PMID 21967765.
- Veggiotti, P; Pera, M. C; Teutonico, F; Brazzo, D; Balottin, U; Tassinari, C. A (2012). "Therapy of encephalopathy with status epilepticus during sleep (ESES/CSWS syndrome): An update". Epileptic Disorders. 14 (1): 1–11. doi:10.1684/epd.2012.0482. PMID 22426353.
- Perucca, Piero; Gilliam, Frank G (2012). "Adverse effects of antiepileptic drugs". The Lancet Neurology. 11 (9): 792–802. doi:10.1016/S1474-4422(12)70153-9. PMID 22832500.
- Guerrini, Renzo; Zaccara, Gaetano; La Marca, Giancarlo; Rosati, Anna (2012). "Safety and Tolerability of Antiepileptic Drug Treatment in Children with Epilepsy" (PDF). Drug Safety. 35 (7): 519–33. doi:10.2165/11630700-000000000-00000. hdl:2158/647763. PMID 22702637.
- Depaulis, Antoine; van Luijtelaar, Gilles (2005). "Genetic models of absence epilepsy in the rat". In Pitkänen, Asla; Schwartzkroin, Philip A.; Moshé, Solomon L. (eds.). Models of Seizures and Epilepsy. Elsevier. pp. 233–48. ISBN 978-0-12-088554-1.