Retro-Diels–Alder reaction
The retro-Diels–Alder reaction (rDA) is the microscopic reverse of the Diels–Alder reaction—the formation of a diene and dienophile from a cyclohexene. It can be accomplished spontaneously with heat, or with acid or base mediation.[1][2]
In principle, it becomes thermodynamically favorable for the Diels–Alder reactions to proceed in the reverse direction if the temperature is high enough. In practice, this reaction generally requires some special structural features in order to proceed at temperatures of synthetic relevance. For instance, the cleavage of cyclohexene to give butadiene and ethylene has been observed, but only at temperatures exceeding 800 K.[3] With an appropriate driving force, however, the Diels–Alder reaction proceeds in reverse under relatively mild conditions, providing diene and dienophile from starting cyclohexene derivatives. As early as 1929, this process was known and applied to the detection of cyclohexadienes, which released ethylene and aromatic compounds after reacting with acetylenes through a Diels–Alder/retro-Diels–Alder sequence.[4] Since then, a variety of substrates have been subject to the rDA, yielding many different dienes and dienophiles. Additionally, conducting the rDA in the presence of a scavenging diene or dienophile has led to the capture of many transient reactive species.[5]
(1)

Mechanism and stereochemistry
Prevailing mechanism
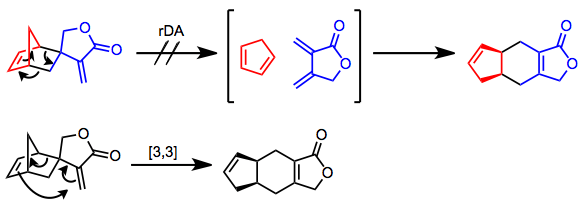
The retro-Diels–Alder reaction proper is the microscopic reverse of the Diels–Alder reaction: a concerted (but not necessarily synchronous), pericyclic, single-step process. Evidence for the retro-Diels–Alder reaction was provided by the observation of endo-exo isomerization of Diels–Alder adducts.[6] It was postulated that at high temperatures, isomerization of kinetic endo adducts to more thermodynamically stable exo products occurred via an rDA/DA sequence. However, such isomerization may take place via a completely intramolecular, [3,3]-sigmatropic (Cope) process. Evidence for the latter was provided by the reaction below—none of the "head-to-head" isomer was obtained, suggesting a fully intramolecular isomerization process.[7]
(2)
Stereochemistry
Like the Diels–Alder reaction, the rDA preserves configuration in the diene and dienophile. Much less is known about the relative rates of reversion of endo and exo adducts, and studies have pointed to no correlation between relative configuration in the cyclohexene starting material and reversion rate.[8]
Scope and limitations
A few rDA reactions occur spontaneously at room temperature because of the high reactivity or volatility of the emitted dienophile. Most, however, require additional thermal or chemical activation. The relative tendencies of a variety of dienes and dienophiles to form via rDA are described below:
Diene: furan, pyrrole > benzene > naphthalene > fulvene > cyclopentadiene > anthracene > butadiene
Dienophile: N2 > CO2 > naphthalene > benzene, nitriles > methacrylate > maleimides > cyclopentadiene, imines, alkenes > alkynes
All-carbon dienophiles
Because the Diels–Alder reaction exchanges two π bonds for two σ bonds, it is intrinsically thermodynamically favored in the forward direction. However, a variety of strategies for overcoming this inherent thermodynamic bias are known. Complexation of Lewis acids to basic functionality in the starting material may induce the retro-Diels–Alder reaction, even in cases when the forward reaction is intramolecular.[9]
(3)
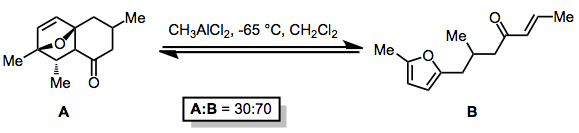
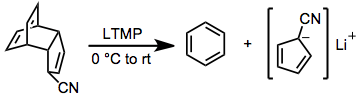
Base mediation can be used to induce rDA in cases when the separated products are less basic than the starting material. This strategy has been used, for instance, to generate aromatic cyclopentadienyl anions from adducts of cyclopentadiene.[10] Strategically placed electron-withdrawing groups in the starting material can render this process essentially irreversible.
(4)
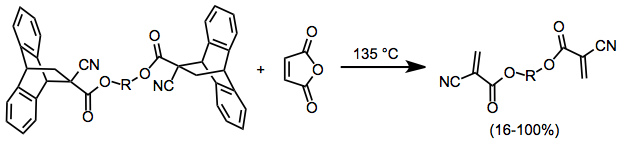
If isolation or reaction of an elusive diene or dienophile is the goal, one of two strategies may be used. Flash vacuum pyrolysis of Diels–Alder adducts synthesized by independent means can provide extremely reactive, short-lived dienophiles (which can then be captured by a unique diene).[11] Alternatively, the rDA reaction may be carried out in the presence of a scavenger. The scavenger reacts with either the diene or (more typically) the dienophile to drive the equilibrium of the retro-DA process toward products. Highly reactive cyanoacrylates may be isolated from Diels–Alder adducts (synthesized independently) with the use of a scavenger.[12]
(5)
Heteroatomic dienophiles
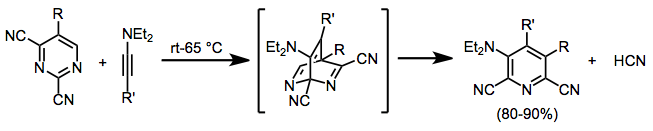
Nitriles may be released in rDA reactions of DA adducts of pyrimidines or pyrazines. The resulting highly substituted pyridines can be difficult to access by other means.[13]
(6)
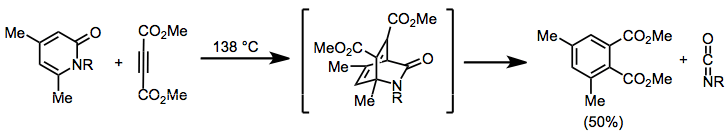
Release of isocyanates from Diels–Alder adducts of pyridones can be used to generate highly substituted aromatic compounds. The isocyanates may be isolated or trapped if they are the desired product.[14]
(7)
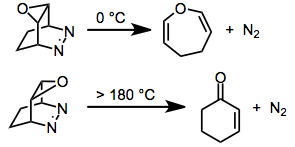
Release of nitrogen from six-membered, cyclic diazenes is common and often spontaneous at room temperature. In this particular example, the epoxide shown undergoes rDA at 0 °C. The isomer with a cis relationship between the diazene and epoxide reacts only after heating to >180 °C.[15]
(8)
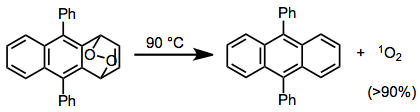
The concerted release of oxygen via rDA results in the formation of singlet oxygen. Very high yields of singlet oxygen result from rDA reactions of some cyclic peroxides—in this example, a greater than 90% yield of singlet oxygen was obtained.[16]
(9)
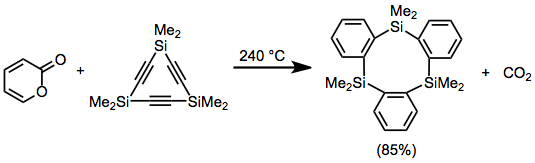
Carbon dioxide is a common dienophile released during rDA reactions. Diels–Alder adducts of alkynes and 2-pyrones can undergo rDA to release carbon dioxide and generate aromatic compounds.[17]
(10)
Experimental conditions and procedure
Typical conditions
Internal energy is the only factor controlling the extent of rDA reactions, and temperature is usually the only variable cited for these reactions. Thus, there are no conditions which can be regarded as "typical." For rDA reactions that afford a volatile product, removal of this product may facilitate the reaction, although most of these reactions (nitrogen- and oxygen-releasing rDA, for instance) are irreversible without any extra inducement.
References
- Rickborn, B. (1998). The Retro–Diels–Alder Reaction. Part I. C–C Dienophiles. Org. React. 52. pp. 1–393. doi:10.1002/0471264180.or052.01. ISBN 978-0471264187.
- Rickborn, B. (1998). The Retro-Diels-Alder Reaction. Part II. Dienophiles with One or More Heteroatoms. Org. React. 53. pp. 223–629. doi:10.1002/0471264180.or053.02. ISBN 978-0471264187.
- Uchiyama, Masao; Tomioka, Tadao; Amano, Akira (2002-05-01). "Thermal Decomposition of Cyclohexene". The Journal of Physical Chemistry. 68 (7): 1878–1881. doi:10.1021/j100789a036.
- Diels, O.; Alder, K.; Stein, G.; Pries, P.; Winckler, H. (1929). "Synthesen in der hydroaromatischen Reihe, VI. Mitteilung, Kurt Alder und Gerhard Stein: Über partiell hydrierte Naphtho- und Anthrachinone mit Wasserstoff in γ- bzw. δ-Stellung. (Mitbearbeitet von Paul Pries und Hans Winckler)". Chem. Ber. 62 (8): 2337. doi:10.1002/cber.19290620872.
- Ichihara, A. (1987). "Retro-Diels-Alder Strategy in Natural Product Synthesis". Synthesis. 1987 (3): 207–222. doi:10.1055/s-1987-27894.
- Alder, K.; Rickert, H. F. (1936). "Zur Kenntnis der Dien-synthese. I. Über eine Methode der direkten Unterscheidung cyclischer Penta- und Hexa-diene". Justus Liebigs Ann. Chem. 524: 180–189. doi:10.1002/jlac.19365240109.
- Haslouin, J.; Rouessac, F. (1977). Bull. Soc. Chim. Fr. Pt. 2: 1242. Missing or empty
|title=(help) - Rye, A. R.; Wege, D. (1974). "Preparation and thermolysis of exo-and endo-Tricyclo[6,2,1,02,7] undeca-3,5,9-triene". Aust. J. Chem. 27 (9): 1943. doi:10.1071/CH9741943.
- Rogers, C.; Keay, B. A. (1991). "Catalytic methylaluminum dichloride: An efficient method for accelerating the intramolecular Diels-Alder reaction of the furan diene" (PDF). Tetrahedron Lett. 32 (45): 6477. doi:10.1016/0040-4039(91)80197-E. hdl:1880/44897.
- Neukam, W.; Grimme, W. (1978). "Anionic (4+2)-cycloreversions leading to the cyanocyclopentadienide ion". Tetrahedron Lett. 19 (25): 2201. doi:10.1016/S0040-4039(01)86845-3.
- Ahmar, M.; Romain, I.; Bloch, R. (1993). "Efficient and highly stereoselective syntheses of enantiomerically enriched C(1)-C(7) subunits of erythronolides". J. Org. Chem. 58 (11): 2953. doi:10.1021/jo00063a009.
- Buck, C. J. J. (1978). "Unequivocal synthesis of bis(2-cyanoacrylate) monomers. I. Via anthracene adducts". Polym. Sci., Polym. Chem. Ed. 16 (10): 2475–2507. Bibcode:1978JPoSA..16.2475B. doi:10.1002/pol.1978.170161007.
- Martin, J. C. J. (1980). "Synthesis of pyridines from dicyanopyrimidines. A diels-alder approach to the c-ring of streptonigrin". Heterocycl. Chem. 17 (5): 1111–1112. doi:10.1002/jhet.5570170554.
- Afarinkia, K.; Vinader, V.; Nelson, T. D.; Posner, G. H. (1992). "Diels-Alder cycloadditions of 2-pyrones and 2-pyridones". Tetrahedron. 48 (42): 9111. doi:10.1016/S0040-4020(01)85607-6.
- Liao, Y.; White, J. B. (1990). "An examination of the affect of the epoxide stereochemistry in the nitrogen extrusion from exo- and endo-6,7-diazo-3-oxotricyclo[3.2.2.02,4]Non-6-ene". Tetrahedron Lett. 31 (36): 5129. doi:10.1016/S0040-4039(00)97822-5.
- Turro, N. J.; Chow, M. F. (1979). "Magnetic field effects on the thermolysis of endoperoxides of aromatic compounds. Correlations with singlet oxygen yield and activation entropies". J. Am. Chem. Soc. 101 (13): 3701. doi:10.1021/ja00507a067.
- Sakurai, H.; Eriyama, Y.; Hosomi, A.; Nakadaira, Y.; Kabuto, C. (1984). "Preparation and reactions of dodecamethyl-3,4,7,8,11,12-hexasilacyclododeca-1,5,9-triyne". Chem. Lett. 13 (4): 595–598. doi:10.1246/cl.1984.595.