Remyelination
Remyelination is the process of propagating oligodendrocyte precursor cells to form oligodendrocytes to create new myelin sheaths on demyelinated axons in the CNS. This is a process naturally regulated in the body and tends to be very efficient in a healthy CNS.[1] The process creates a thinner myelin sheath than normal, but it helps to protect the axon from further damage, from overall degeneration, and proves to increase conductance once again. The processes underlying remyelination are under investigation in the hope of finding treatments for Demyelinating diseases, such as multiple sclerosis.
Function
Remyelination is activated and regulated by a variety of factors surrounding lesion sites that control the migration and differentiation of Oligodendrocyte Precursor Cells. Remyelination looks different from developmental myelination in the structure of the myelin formed. Reasons for this are unclear, but proper function of the axon is restored regardless. Perhaps of most interest are the inhibition and promotion factors of this physiological process. One way this process can be traced is by following different protein activation sequences which have shown just how quickly remyelination begins after injury (within a few of days).[2]
Characteristics of remyelinated axons
The most notable evidence that remyelination has taken place on an axon is its thin myelin sheath created by an oligodendrocyte, though the reason why the new myelin sheath is thinner remains unclear. This can be quantified in the g-ratio, the ratio between the diameter of the axon itself to the outer diameter of the myelinated fiber. Remyelinated axons tend to have values closer to 1, indicating a thinner myelin sheath than those myelinated naturally. The g-ratio differences are less apparent on smaller axons.[1]
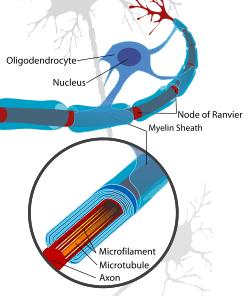
The thinner myelin not only restores protection of the axon from degradation,[3] but also restores a faster conduction velocity. The conduction velocity, however, is not as strong as naturally myelinated axons and the Nodes of Ranvier are inclined to be wider which results in less coverage in the axon by myelin than what is natural.[4]
OPC involvement
Oligodendrocyte Precursor Cells, or OPC's, are the main cells responsible for the remyelination of demyelinated axons. There are two physiological changes that must occur to OPC's for remyelination to occur.[1] Once a signal is sent that remyelination is needed, OPC's will first migrate to damaged axon. This process may be signaled or enhanced by microglia or astrocytes at the injured axon site that stimulate migratory OPC pathways [1] From there the cells must differentiate from being progenitors to being pre-oligodendrocytes, then premyelinating oligodendrocytes, and finally mature oligodendrocytes.[4] These oligodendrocytes can then wrap damaged axons with new myelin sheaths. This process of differentiation through several phases has many involved and direct pathways and factors necessary for the completion of this process. It is easy to completely stop remyelination with the failure of a number of pathways.
Propagation factors
One of the difficulties of studying remyelination is the variety of factors that play a role in differentiating oligodendrocyte progenitors. While some factors promote and others inhibit, still some factors that are known to be involved are yet not understood enough to know whether it promotes, inhibits, or does both. Many factors are poorly understood and subject to much change as research is done.
Cytokines and chemokines
Cytokines mediate inflammatory responses that promote pathogen and debris clearance so that further tissue damage is avoided.[1][4][5] Too much can mean cell death but failure to propagate cytokines at all in remyelination results in a lack of debris clearance at a damaged axon site; this buildup of myelin and oligodendrocyte debris has been shown to inhibit the differentiation of Oligodendrocyte Precursor Cells.[1] Specifically, cytokines promote TNFR2 and eventually TNF-alpha which plays a key role in OPC differentiation.[5]
It has also been shown that chemokines are involved in guiding immune cells to sites of axon lesions to facilitate inflammation and debris clearance as well as possibly guiding OPCs migration to lesion sites. So then, chemokines are directly involved with both migration and differentiation of OPCs.[5] The specific chemokines involved with each of these two processes is known: CXCL12 is related to migration and differentiation is increased with an increase in CXCR7 and a decrease in CXCR4.[5] In certain demyelinating diseases CXCL12 has been shown to be decreased, possibly playing a role in demyelination failure. Still much is to be researched in this field, as certain chemokines like CXCR2 plays a role in inflammation and repair but in an unknown manner over much controversy.[5]
Signaling pathways
LINGO1, a cell receptor, has been proposed to be involved in the regulation of remyelination. It is thought to inhibit not only axon regeneration but also regulate oligodendrocyte maturation by inhibiting OPC differentiation. Animal studies suggest that when a LINGO1 is inhibited,[6] OPC differentiation and thus remyelination can be promoted at demyelinated sites. LINGO1 gene expression is also known to activate RhoA which may also play a part in inhibition.[1][4][5] Myelin debris build up might be responsible for the promotion of the LINGO1 signalling and overall inhibition.[4][7][8]
The Notch-1 receptor pathway is another pathway that inhibits the differentiation of OPCs.[4] When the ligands Jagged1 and Delta, produced by axons, neurons, and astrocytes, are stimulated and bind to the membrane, oligodendrocyte maturation is inhibited. This pathway may also be facilitating migration despite its differentiation inhibition.[5] In some experiments, altering the pathway so that differentiation is increased caused a decrease in the proliferation of OPCs.[9] There may be other ligands that have either promoting or inhibiting effects when attached to the Notch-1 receptor.[1][9]
The Wnt-β-Catelin pathway has been shown to also inhibit remyelination when it is dysregulated in the body. Demyelinating diseases have been shown to cause this dysregulation. Possible genes involved inside this pathway are TCF4 and OLIG2 which are both expressed in high amounts in areas where remyelination has failed from demyelinating diseases.[4][10]
Transcription factors
Gene expression may be the most important factor in understanding remyelination and can hold the key to understanding how to treat demyelinating diseases. OLIG1 has been shown to be critical in developmental myelination and may also be important in remyelination.[5] OLIG2 and TRF4 have also been shown to be important especially in the Wnt-β-Catenin Pathway, most likely in inhibiting remyelination. NKX2-2 is a gene coding for a protein that may increase the number of OPCs in low amounts, possibly working with OLIG2 in some way to differentiate OPCs to mature oligodendrocytes.[5] As more genes involved in remyelination are found and cross linked more will be understood about promotion and inhibition.
Androgen receptor (AR) and testosterone
In a mouse model, it has been shown that testosterone, acting through the AR, is important in remyelination by oligodendrocytes.[11][12] Those same authors note that the AR evolved from a duplicated gene coincidentally with the development of myelin in jawed vertebrates.
Other factors
It is known that as age increases there's a decrease in the efficiency (both the speed and magnitude) of remyelination at demyelinated axons. This is probably linked with down regulation of certain expressed genes with increased age. The research of this is particularly important with the elderly whose myelin and axons are more prone to be degenerated in the CNS.[1][13]
Class 3 semaphorins (SEMA3s), originally identified as axon guidance molecules, play a role in remyelination. For instance, SEMA3s modulate the recruitment of oligodendrocyte precursor cells and their differentiation into oligodendrocytes. In addition, SEMA3a is known to repel Schwann cells.[14]
Growth Factors are active polypeptides that control differentiation and biological growth in responsive cells. They have been shown to have a prominent role. Due to the wide variety of these factors it is difficult to study specifically but understanding can be big in treating demyelinating diseases. Some of the factors being researched are EGF (which is known to enhance myelination), IGF-1, PDGF, and FGF [5]
Toll-Like receptors are also involved in remyelination, most likely inhibiting remyelination and OPC differentiation. There are a variety of types of these receptors, but a majority of them tend to increase, especially in the chronic stages of demyelinating diseases, suggesting that they may be involved with remyelination failure.[4][5]
MicroRNA is not well understood but may play a minor or major role in remyelination. MicroRNA may have a role in reduction of CD47 which promotes phagocytosis of myelin.[5] Certain microRNAs have been shown to promote OPC differentiation by their involvement and maintenance of genes that keep OPCs undifferentiated.[15]
Disease treatment
Understanding completely the inhibiting and promoting factors of OPCs seems to be the key in battling demyelinating diseases such as multiple sclerosis that cause remyelination to fail.[2] Not only are the inhibition factors being looked at as ways to stop remyelination failure, but promotion factors are being looked at to facilitate remyelination in the face of inhibited processes. Stem cell research is also ongoing in seeing how to differentiate neural stem cells into mature oligodendrocytes that will activate at demyelinated sites.[5] Looking at the known factors of developmental myelination may also translate well into remyelination promotion.[5]
Multiple sclerosis
Multiple sclerosis, or MS, is the most prominent of the demyelinating diseases, affecting at least 30 in 100000 people worldwide on average. The ratio is much higher than that in certain areas of the world. While the early stages of multiple sclerosis are less discernible, the chronic stages can greatly reduce an individual's quality of life by limiting motor function. The demyelinating disease attacks the myelin of axons in the central nervous system through autoimmune defects. While remyelination is very efficient in the early stages of multiple sclerosis, it causes remyelination to fail in the more chronic stages.[1] As axons are left bare, without myelin, their conduction velocity goes down due to a lack in increased potential between the Nodes of Ranvier. Not only does conduction go down, but a naked axon is also much more likely to degrade completely, resulting in complete loss of function for certain motor functions. The loss of axons because of lack of protection is what makes MS so debilitating. Degradation is considered to be worse than the effects of demyelination.[3] Once an axon is degenerated, it cannot regenerate like myelin, thus making research to promote remyelination that much more important. MS is more severe in some people than others, most likely from their family genetics and the way that genes are expressed within them.[4] The overall cause for multiple sclerosis itself is completely unknown. Altering important pathways in OPC differentiation such as Notch-1, Wnt, and LINGO1 may prove to be a possible treatment for this disease.[1][5][9] Using antibodies to halt or promote certain parts of these pathways could be possible therapies to help increase OPC differentiation. As pathways are better understood, different parts of the pathways can be singled out as possible therapeutic areas to promote remyelination.
Future research
Still much is not understood concerning remyelination. New pathways are being discovered constantly in the areas of gene regulation, antibody use as antagonists, and promotion of stem cells to differentiate. There are many regulation factors, such as Lingo-1, Olig-1, Id2, Id4, Hes5, and Sox6, that are not very well understood in their role that may hold the key to developing new treatments for demyelinating diseases.[1][15] One of the biggest difficulties of studying demyelinating diseases and thus remyelination is that it takes place in the central nervous system. Studying remyelination most thoroughly would involve unethical and invasive experiments and observation on the human brain and spinal cord.[1][5] Because of this, scientists are limited to studying patients with demylinating diseases after they have died. It is nearly impossible to discern what exactly happened through the progress of the diseased person because most persons die in the chronic stages of their demyelinating disease. The other method of studying demyelinating diseases is using animals. Specifically, rats and mice are commonly used to investigate remyelination. The most commonly employed models rely on toxins that are used to generate focal or generalised demyelination in the CNS.[16][17] Unlike in MS-mimmicking animal models, such as Experimental autoimmune encephalomyelitis, or EAE, toxin models allow for precisely controlled demyelination. EAE is induced by immunologically sensitising animals to myelin components. Although EAE is not the same as MS, it reproduces a similar environment and many of the same effects.[1]
References
- Franklin, RJM; C. Ffrench-Constant (November 2008). "Remyelination in the central nervous system (CNS): from biology to therapy". Nature Reviews Neuroscience. 9 (11): 839–855. doi:10.1038/nrn2480. PMID 18931697.
- Lindner, M.; Heine, S.; Haastert, K.; Garde, N.; Fokuhl, J.; Linsmeier, F.; Grothe, C.; Baumgärtner, W.; Stangel, M. (24 October 2007). "Sequential myelin protein expression during remyelination reveals fast and efficient repair after central nervous system demyelination". Neuropathology and Applied Neurobiology. 34 (1): 105–114. doi:10.1111/j.1365-2990.2007.00879.x. PMID 17961136.
- Irvine, K. A.; Blakemore, W. F. (29 January 2008). "Remyelination protects axons from demyelination-associated axon degeneration". Brain. 131 (6): 1464–1477. doi:10.1093/brain/awn080. PMID 18490361.
- Hanafy, Khalid A.; Sloane, Jacob A. (1 December 2011). "Regulation of remyelination in multiple sclerosis". FEBS Letters. 585 (23): 3821–3828. doi:10.1016/j.febslet.2011.03.048. PMID 21443876.
- Patel, Jigisha R.; Klein, Robyn S. (1 December 2011). "Mediators of oligodendrocyte differentiation during remyelination". FEBS Letters. 585 (23): 3730–3737. doi:10.1016/j.febslet.2011.04.037. PMC 3158966. PMID 21539842.
- Mi, Sha; Miller, Robert H.; Tang, Wei; Lee, Xinhua; Hu, Bing; Wu, Wutain; Zhang, Yiping; Shields, Christopher B.; Zhang, Yongjie; Miklasz, Steven; Shea, Diana; Mason, Jeff; Franklin, Robin J. M.; Ji, Benxiu; Shao, Zhaohui; Chédotal, Alain; Bernard, Frederic; Roulois, Aude; Xu, Janfeng; Jung, Vincent; Pepinsky, Blake (1 March 2009). "Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells". Annals of Neurology. 65 (3): 304–315. doi:10.1002/ana.21581. PMID 19334062.
- Kotter, Mark R.; Li, Wen-Wu; Zhao, Chao; Franklin, Robin J. M. (2006-01-04). "Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation". The Journal of Neuroscience. 26 (1): 328–332. doi:10.1523/JNEUROSCI.2615-05.2006. ISSN 0270-6474. PMC 6674302. PMID 16399703.
- Baer, Alexandra S.; Syed, Yasir A.; Kang, Sung Ung; Mitteregger, Dieter; Vig, Raluca; ffrench-Constant, Charles; Franklin, Robin J. M.; Altmann, Friedrich; Lubec, Gert (2009-02-01). "Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling". Brain. 132 (2): 465–481. doi:10.1093/brain/awn334. ISSN 0006-8950. PMC 2640211. PMID 19208690.
- Zhang, Yueting; et al. (November 10, 2009). "Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination". PNAS. 106 (45): 19162–19167. doi:10.1073/pnas.0902834106. PMC 2776461. PMID 19855010.
- Fancy, S. P.J.; Baranzini, S. E.; Zhao, C.; Yuk, D.-I.; Irvine, K.-A.; Kaing, S.; Sanai, N.; Franklin, R. J.M.; Rowitch, D. H. (10 June 2009). "Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS". Genes & Development. 23 (13): 1571–1585. doi:10.1101/gad.1806309. PMC 2704469. PMID 19515974.
- Bielecki, B; et al. (2016). "Unexpected central role of the androgen receptor in the spontaneous regeneration of myelin". PNAS USA. 113 (51): 14829–14834. doi:10.1073/pnas.1614826113. PMC 5187716. PMID 27930320.
- Hussain, Rashad; Ghoumari, Abdel M.; Bielecki, Bartosz; Steibel, Jérôme; Boehm, Nelly; Liere, Philippe; Macklin, Wendy B.; Kumar, Narender; Habert, René; Mhaouty-Kodja, Sakina; Tronche, François; Sitruk-Ware, Regine; Schumacher, Michael; Ghandour, M. Said (2013-01-01). "The neural androgen receptor: a therapeutic target for myelin repair in chronic demyelination" (PDF). Brain. 136 (1): 132–146. doi:10.1093/brain/aws284. ISSN 0006-8950. PMC 4572509. PMID 23365095.
- Shen, Siming; Sandoval, Juan; Swiss, Victoria A; Li, Jiadong; Dupree, Jeff; Franklin, Robin J M; Casaccia-Bonnefil, Patrizia (24 June 2008). "Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency". Nature Neuroscience. 11 (9): 1024–1034. doi:10.1038/nn.2172. PMC 2656679. PMID 19160500.
- Mecollari, V; Nieuwenhuis, B; Verhaagen, J (2014). "A perspective on the role of class III semaphorin signaling in central nervous system trauma". Frontiers in Cellular Neuroscience. 8: 328. doi:10.3389/fncel.2014.00328. PMC 4209881. PMID 25386118.
- Emery, B. (4 November 2010). "Regulation of Oligodendrocyte Differentiation and Myelination". Science. 330 (6005): 779–782. doi:10.1126/science.1190927. PMID 21051629.
- Blakemore, WF (1972). "Observations on oligodendrocyte degeneration, the resolution of status spongiosus and remyelination in cuprizone intoxication in mice". J. Neurocytol. 1 (4): 413–26. doi:10.1007/bf01102943. PMID 8530973.
- Woodruff, Rachel H.; Franklin, Robin J.M. (1999-02-01). "Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: A comparative study". Glia. 25 (3): 216–228. doi:10.1002/(sici)1098-1136(19990201)25:3<216::aid-glia2>3.0.co;2-l. ISSN 1098-1136. PMID 9932868.