Reduced representation bisulfite sequencing
Reduced representation bisulfite sequencing (RRBS) is an efficient and high-throughput technique for analyzing the genome-wide methylation profiles on a single nucleotide level. It combines restriction enzymes and bisulfite sequencing to enrich for areas of the genome with a high CpG content. Due to the high cost and depth of sequencing to analyze methylation status in the entire genome, Meissner et al. developed this technique in 2005 to reduce the amount of nucleotides required to sequence to 1% of the genome.[1] The fragments that comprise the reduced genome still include the majority of promoters, as well as regions such as repeated sequences that are difficult to profile using conventional bisulfite sequencing approaches.[2]
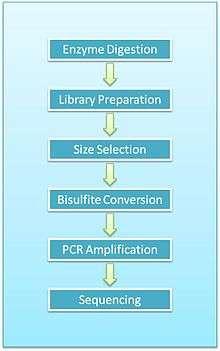
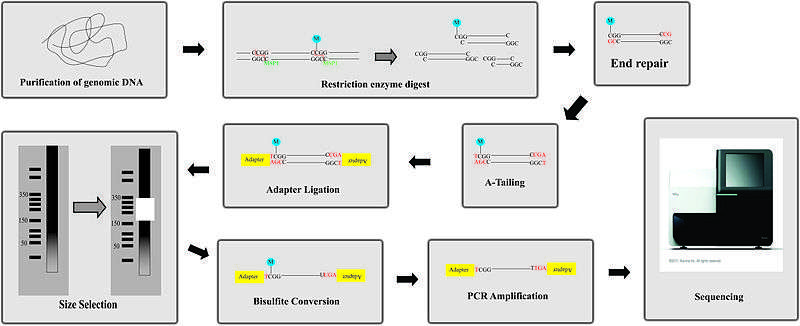
Overview of protocol
- Enzyme digestion: First, genomic DNA is digested using a methylation-insensitive restriction enzyme. It is integral for the enzymes to not be influenced by the methylation status of the CpGs (sites within the genome where a cytosine is next to a guanine) as this allows for the digestion of both methylated and unmethylated areas. MspI is commonly used. This enzyme targets 5’CCGG3’ sequences and cleaves the phosphodiester bonds upstream of CpG dinucleotide. When using this particular enzyme, each fragment has a CpG at each end. This digestion results in DNA fragments of various sizes.
- End repair and A-tailing: Due to the nature of how MspI cleaves double stranded DNA, this reaction results in strands with sticky ends. End repair is necessary to fill in the 3’ terminal of the ends of the strands. The next step is adding an extra adenosine to both the plus and minus strands. This is referred to as A-Tailing and is necessary for adapter ligation in the subsequent step. End repair and A-Tailing is done within the same reactions, with dCTP, dGTP and dATP deoxyribonucleotides. To increase the efficiency of A tailing, the dATPs are added in excess in this reaction.
- Sequence adapters: Methylated sequence adapters are ligated to the DNA fragments. The methylated adapter oligonucleotides have all cytosines replaced with 5’methyl-cytosines to prevent deamination of these cytosines in the bisulfite conversion reaction. To sequence reactions using Illumina sequencers, sequence adapters hybridize to the adapters on the flow cell.
- Fragment purification: The desired size of fragments is selected for purification. Different sizes of the fragments are separated using gel electrophoresis and are purified using gel excising. According to Gu et al., DNA fragments of 40-220 base pair are representative of the majority of promoter sequences and CpG islands[2]
- Bisulfite conversion: The DNA fragments are then bisulfite converted, which is a process that deaminates unmethylated cytosine into a uracil. The methylated cytosines remain unchanged, due to the methyl group protecting them from the reaction.
- PCR amplification: The bisulfite converted DNA is then amplified using PCR with primers that are complementary to the sequence adapters.
- PCR purification: Before sequencing, the PCR product must be free of unused reaction reagents such as unincorporated dNTPs or salts. Thus, a step for PCR purification is required. This can be done by running another electrophoresis gel or by using kits designed specifically for PCR purification.
- Sequencing: The fragments are then sequenced. When RRBS was first developed, Sanger sequencing was initially used. Now, next generation sequencing approaches are used. For Illumina sequencing, 36-base single-end sequencing reads are most commonly performed.
- Sequence alignment and analysis: Due to the unique properties of RRBS, special software is needed for alignment and analysis.[3] Using MspI to digest genomic DNA results in fragments that always start with a C (if the cytosine is methylated) or a T (if a cytosine was not methylated and was converted to a uracil in the bisulfite conversion reaction). This results in a non-random base pair composition. Additionally, the base composition is skewed due to the biased frequencies of C and T within the samples. Various software for alignment and analysis is available, such as Maq, BS Seeker, Bismark or BSMAP. Alignment to a reference genome allows the programs to identify base pairs within the genome that are methylated.
Advantages
Enrichment of CpGs
RRBS uniquely uses a specific restriction enzyme to enrich for CpGs. MspI digestion, or any restriction enzyme that recognizes CpG's and cuts them, produces only fragments with CG’s at the end.[2] This approach enriches for CpG regions of the genome, so it can decrease the amount of sequencing required as well as decrease the cost.[2] This technique is cost-effective especially when focusing on common CpG regions.
Low sample input
Only a low sample concentration, between 10-300 ng, is required for accurate data analysis.[2] This technique can be employed when there is a lack of precious sample. Another positive aspect is that fresh or live samples are not required. Formalin-fixed and paraffin-embedded inputs can also be used.[2]
Limitations
Restriction enzyme
In the specific protocol steps, there are also some limitations. MspI digestion covers the majority, but not all the CG regions in the genome.[2] Some CpG’s are missed. Missing CpG’s can also occur since this protocol is only a representative sampling of the genome.[2] Some regions thus have lower coverage. Other variations of this protocol use alternative enzymes.[4]
PCR
During the PCR portion of the protocol, a non-proofreading polymerase must be used as a proof-reading enzyme would stop at uracil residues found in the ssDNA template.[1] Using a polymerase that does not proof-read can also lead to increase PCR sequencing errors.[1]
Bisulfite sequencing
Bisulfite sequencing only converts single-stranded DNA (ssDNA). Complete bisulfite conversion requires thorough denaturation and absence of re-annealed double stranded DNA (dsDNA).[1] Easy protocol steps have been shown to drive complete denaturation. Ensuring the usage of small fragments via shearing or digestion, fresh reagents, and sufficient denaturing time is crucial for complete denaturing [5] Another suggested technique is to carry out the bisulfite reaction at 95 °C although DNA degradation also occurs at high temperatures.[5] In the first hour of bisulfite reaction, it is predicted that less than 90% of the sample DNA is lost to degradation [6] A balance between high temperature and low temperature is required to ensure complete denaturation and decreased DNA degradation. Usage of reagents, like urea, that prevent dsDNA from forming can also be employed.[5] With contamination of dsDNA, it can be difficult to accurately computate the data.[1] When an unconverted cytosine is observed, it is challenging to differentiate between lack of methylation and an artifact.[1]
Significance
The significance of this technique is it allows for the sequencing of methylated areas that can't be properly profiled using conventional bisulfite sequencing techniques. Current sequencing technologies are limited in regards to profiling areas of repeated sequences.[7] This is unfortunate in regards to methylation studies, as these repeated sequences often contain methylated cytosines. This is especially limiting for studies involving profiling cancer genomes, as a loss of methylation in this repeated sequences is observed in many cancer types.[8] RRBS eliminates the problems encountered due to these large areas of repeated sequences and thus lets these regions be more fully annotated.
Applications
Methylomes in cancer genomics
Aberrant methylation has been observed in cancer.[9] In cancer, hypermethylation as well as hypomethylation has been seen in tumors.[9] Since RRBS is highly sensitive, this technique can be used to quickly look at aberrant methylation in cancer.[10] If samples from the patient's tumor and normal cells can be obtained, a comparison between these two cell types can be observed.[2][10] A profile of the overall methylation can be produced quite rapidly.[2] This technique can rapidly determine the overall methylation status of cancer genomes which is cost and time effective.
Methylation states in development
Stage-specific changes can be observed in all living organisms. Modifications in overall methylation levels via reduced representation bisulfite sequencing can be useful in developmental biology.
Comparison with other techniques
Results compared between RRBS and MethylC-seq are highly concordant with one another.[11] Naturally, MethylC-seq has a greater genome-wide coverage of CpGs compared to RRBS, but RRBS has a greater coverage on CpG islands.[11] One of the other most commonly used techniques for profling methylation is MeDiP-Seq. This technique is done by immunopreciptiation of methylated cytosines and subsequent sequencing.[11] RRBS has a greater resolution compared to this technique, as MeDip-Seq is limited to 150 base pairs compared the one nucleotide resolution of RRBS.[11] Bisulfite methods, such as used by RRBS, were also found more accurate than enrichment based, such as MeDip-Seq.[7] The data obtained on RRBS and the Illumina Infinium methylation are highly comparable, with a Pearson correlation of 0.92.[7] The data for both platforms are also directly comparable as both use an absolute measurement of DNA.
References
- Alexander Meissner, Andreas Gnirke, George W. Bell, Bernard Ramsahoye, Eric S. Lander and Rudolf Jaenisch. 2005. "Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis". Nucleic Acids Res. 33(18):5868-77
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. 2011. "Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling." Nat Protoc. 6(4):468-81. doi:10.1038/nprot.2010.190.
- Chatterjee A, Rodger EJ, Stockwell PA, Weeks RJ, Morison IM. 2012. "Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries." J Biomed Biotechnol. 2012:741542. doi:10.1155/2012/741542
- Trucchi, Emiliano; Mazzarella, Anna B.; Gilfillan, Gregor D.; Lorenzo, Maria T.; Schönswetter, Peter; Paun, Ovidiu (April 2016). "BsRADseq: screening DNA methylation in natural populations of non-model species". Molecular Ecology. 25 (8): 1697–1713. doi:10.1111/mec.13550. PMC 4949719. PMID 26818626.
- Warnercke, P.M. Stirzaker, C., Song, J., Grunau, C., Melki, J.R., Clark, S.J. 2002. "Identification and resolution of artifacts in bisulphite sequencing." Methods. 27: 101-107.
- Grunau,C., Clark,S.J. and Rosenthal,A. 2001. "Bisulfite genomic sequencing: systematic investigation of critical experimental parameters." Nucleic Acids Res., 29, E65–65.
- Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, Jäger N, Gnirke A, Stunnenberg HG, Meissner A. 2010. "Quantitative comparison of genome-wide DNA methylation mapping technologies." Nat Biotechnol. 28(10):1106-14. doi:10.1038/nbt.1681
- Ehrlich M. 2009. "DNA hypomethylation in cancer cells". Epigenomics. 1(2):239-59. doi:10.2217/epi.09.33.
- Veersteeg, R. 1997. Aberrant methylation in cancer. American Journal of Human Genetics. 60:751-754.
- Smith, Z.D., Gu, H., Bock, C., Gnike, A., & Meissner, A. 2009. High-throughput bisulfite sequencing in mammalian genomes. Methods. 48: 226-232.
- Harris, Alan et al. 2010. “Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications” Nature Biotechnology 28:1097-1105.