Phred quality score
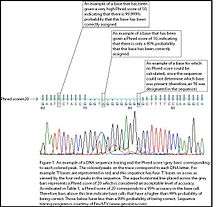
A Phred quality score is a measure of the quality of the identification of the nucleobases generated by automated DNA sequencing.[1][2] It was originally developed for Phred base calling to help in the automation of DNA sequencing in the Human Genome Project. Phred quality scores are assigned to each nucleotide base call in automated sequencer traces.[3][2] The FASTQ format encodes phred scores as ASCII characters alongside the read sequences. Phred quality scores have become widely accepted to characterize the quality of DNA sequences, and can be used to compare the efficacy of different sequencing methods. Perhaps the most important use of Phred quality scores is the automatic determination of accurate, quality-based consensus sequences.
Definition
Phred quality scores are defined as a property which is logarithmically related to the base-calling error probabilities .[2]



or

For example, if Phred assigns a quality score of 30 to a base, the chances that this base is called incorrectly are 1 in 1000.
| Phred Quality Score | Probability of incorrect base call | Base call accuracy |
|---|---|---|
| 10 | 1 in 10 | 90% |
| 20 | 1 in 100 | 99% |
| 30 | 1 in 1000 | 99.9% |
| 40 | 1 in 10,000 | 99.99% |
| 50 | 1 in 100,000 | 99.999% |
| 60 | 1 in 1,000,000 | 99.9999% |
The phred quality score is the negative ratio of the error probability to the reference level of expressed in Decibel (dB).

History
The idea of sequence quality scores can be traced back to the original description of the SCF file format by Staden's group in 1992.[4] In 1995, Bonfield and Staden proposed a method to use base-specific quality scores to improve the accuracy of consensus sequences in DNA sequencing projects.[5]
However, early attempts to develop base-specific quality scores[6][7] had only limited success.
The first program to develop accurate and powerful base-specific quality scores was the program Phred. Phred was able to calculate highly accurate quality scores that were logarithmically linked to the error probabilities. Phred was quickly adopted by all the major genome sequencing centers as well as many other laboratories; the vast majority of the DNA sequences produced during the Human Genome Project were processed with Phred.
After Phred quality scores became the required standard in DNA sequencing, other manufacturers of DNA sequencing instruments, including Li-Cor and ABI, developed similar quality scoring metrics for their base calling software.[8]
Methods
Phred's approach to base calling and calculating quality scores was outlined by Ewing et al.. To determine quality scores, Phred first calculates several parameters related to peak shape and peak resolution at each base. Phred then uses these parameters to look up a corresponding quality score in huge lookup tables. These lookup tables were generated from sequence traces where the correct sequence was known, and are hard coded in Phred; different lookup tables are used for different sequencing chemistries and machines. An evaluation of the accuracy of Phred quality scores for a number of variations in sequencing chemistry and instrumentation showed that Phred quality scores are highly accurate.[9]
Phred was originally developed for "slab gel" sequencing machines like the ABI373. When originally developed, Phred had a lower base calling error rate than the manufacturer's base calling software, which also did not provide quality scores. However, Phred was only partially adapted to the capillary DNA sequencers that became popular later. In contrast, instrument manufacturers like ABI continued to adapt their base calling software changes in sequencing chemistry, and have included the ability to create Phred-like quality scores. Therefore, the need to use Phred for base calling of DNA sequencing traces has diminished, and using the manufacturer's current software versions can often give more accurate results.
Applications
Phred quality scores are used for assessment of sequence quality, recognition and removal of low-quality sequence (end clipping), and determination of accurate consensus sequences.
Originally, Phred quality scores were primarily used by the sequence assembly program Phrap. Phrap was routinely used in some of the largest sequencing projects in the Human Genome Sequencing Project and is currently one of the most widely used DNA sequence assembly programs in the biotech industry. Phrap uses Phred quality scores to determine highly accurate consensus sequences and to estimate the quality of the consensus sequences. Phrap also uses Phred quality scores to estimate whether discrepancies between two overlapping sequences are more likely to arise from random errors, or from different copies of a repeated sequence.
Within the Human Genome Project, the most important use of Phred quality scores was for automatic determination of consensus sequences. Before Phred and Phrap, scientists had to carefully look at discrepancies between overlapping DNA fragments; often, this involved manual determination of the highest-quality sequence, and manual editing of any errors. Phrap's use of Phred quality scores effectively automated finding the highest-quality consensus sequence; in most cases, this completely circumvents the need for any manual editing. As a result, the estimated error rate in assemblies that were created automatically with Phred and Phrap is typically substantially lower than the error rate of manually edited sequence.
In 2009, many commonly used software packages make use of Phred quality scores, albeit to a different extent. Programs like Sequencher use quality scores for display, end clipping, and consensus determination; other programs like CodonCode Aligner also implement quality-based consensus methods.
Compression
Quality scores are normally stored together with the nucleotide sequence in the widely accepted FASTQ format. They account for about half of the required disk space in the FASTQ format (before compression), and therefore the compression of the quality values can significantly reduce storage requirements and speed up analysis and transmission of sequencing data. Both lossless and lossy compression are recently being considered in the literature. For example, the algorithm QualComp [10] performs lossy compression with a rate (number of bits per quality value) specified by the user. Based on rate-distortion theory results, it allocates the number of bits so as to minimize the MSE (mean squared error) between the original (uncompressed) and the reconstructed (after compression) quality values. Other algorithms for compression of quality values include SCALCE,[11] Fastqz[12] and more recently QVZ,[13] AQUa[14] and the MPEG-G standard, that is currently under development by the MPEG standardisation working group. Both are lossless compression algorithms that provide an optional controlled lossy transformation approach. For example, SCALCE reduces the alphabet size based on the observation that “neighboring” quality values are similar in general.
References
- Ewing B; Hillier L; Wendl MC; Green P. (1998). "Base-calling of automated sequencer traces using phred. I. Accuracy assessment". Genome Research. 8 (3): 175–185. doi:10.1101/gr.8.3.175. PMID 9521921.
- Ewing B, Green P (1998). "Base-calling of automated sequencer traces using phred. II. Error probabilities". Genome Research. 8 (3): 186–194. doi:10.1101/gr.8.3.186. PMID 9521922.
- Ewing B, Hillier L, Wendl MC, Green P (1998). "Base-calling of automated sequencer traces using phred. I. Accuracy assessment". Genome Research. 8 (3): 175–185. doi:10.1101/gr.8.3.175. PMID 9521921.CS1 maint: multiple names: authors list (link)
- Dear S, Staden R (1992). "A standard file format for data from DNA sequencing instruments". DNA Sequence. 3 (2): 107–110. doi:10.3109/10425179209034003. PMID 1457811.
- Bonfield JK, Staden R (25 Apr 1995). "The application of numerical estimates of base calling accuracy to DNA sequencing projects". Nucleic Acids Research. 23 (8): 1406–1410. doi:10.1093/nar/23.8.1406. PMC 306869. PMID 7753633.
- Churchill GA, Waterman MS (Sep 1992). "The accuracy of DNA sequences: estimating sequence quality". Genomics. 14 (1): 89–98. doi:10.1016/S0888-7543(05)80288-5. hdl:1813/31678. PMID 1358801.
- Lawrence CB, Solovyev VV (1994). "Assignment of position-specific error probability to primary DNA sequence data". Nucleic Acids Research. 22 (7): 1272–1280. doi:10.1093/nar/22.7.1272. PMC 523653. PMID 8165143.
- "Life Technologies - US" (PDF).
- Richterich P (1998). "Estimation of errors in "raw" DNA sequences: a validation study". Genome Research. 8 (3): 251–259. doi:10.1101/gr.8.3.251. PMC 310698. PMID 9521928.
- Ochoa, Idoia; Asnani, Himanshu; Bharadia, Dinesh; Chowdhury, Mainak; Weissman, Tsachy; Yona, Golan (2013). "Qual Comp: A new lossy compressor for quality scores based on rate distortion theory". BMC Bioinformatics. 14: 187. doi:10.1186/1471-2105-14-187. PMC 3698011. PMID 23758828.
- Hach, F; Numanagic, I; Alkan, C; Sahinalp, S. C. (2012). "SCALCE: Boosting sequence compression algorithms using locally consistent encoding". Bioinformatics. 28 (23): 3051–3057. doi:10.1093/bioinformatics/bts593. PMC 3509486. PMID 23047557.
- "fastqz - FASTQ compressor".
- Malysa, Greg; Hernaez, Mikel; Ochoa, Idoia; Rao, Milind; Ganesan, Karthik; Weissman, Tsachy (2015-10-01). "QVZ: lossy compression of quality values". Bioinformatics. 31 (19): 3122–3129. doi:10.1093/bioinformatics/btv330. ISSN 1367-4803. PMC 5856090. PMID 26026138.
- Paridaens, Tom; Van Wallendael, Glenn; De Neve, Wesley; Lambert, Peter (2018). "AQUa: an adaptive framework for compression of sequencing quality scores with random access functionality". Bioinformatics. 34 (3): 425–433. doi:10.1093/bioinformatics/btx607. PMID 29028894.
External links
- Long Reads with the KB Basecaller Comparison of Phred accuracy with a competing program, ABI's KB Basecaller
- The Laboratory of Phil Green Phrap's homepage.