Photooxygenation
A photooxygenation is a light-induced oxidation reaction in which molecular oxygen is incorporated into the product(s).[1][2] Initial research interest in photooxygenation reactions arose from Oscar Raab's observations in 1900 that the combination of light, oxygen and photosensitizers is highly toxic to cells.[3] Early studies of photooxygenation focused on oxidative damage to DNA and amino acids,[2] but recent research has led to the application of photooxygenation in organic synthesis and photodynamic therapy.[4]
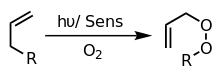
Photooxygenation reactions are initiated by a photosensitizer, which is a molecule that enters an excited state when exposed to light of a specific wavelength (e.g. dyes and pigments). The excited sensitizer then reacts with either a substrate or ground state molecular oxygen, starting a cascade of energy transfers that ultimately result in an oxygenated molecule. Consequently, photooxygenation reactions are categorized by the type and order of these intermediates (as type I, type II, or type III[5] reactions).[2][3]
Background
Terminology
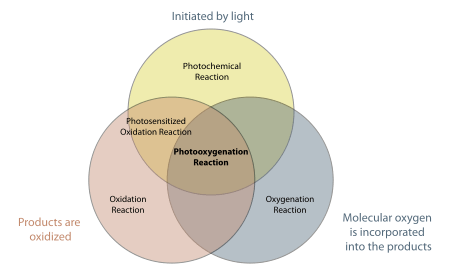
Photooxygenation reactions are easily confused with a number of processes baring similar names (i.e. photosensitized oxidation). Clear distinctions can be made based on three attributes: oxidation, the involvement of light, and the incorporation of molecular oxygen into the products:
Sensitizers
Sensitizers (denoted "Sens") are compounds, such as fluorescein dyes, methylene blue, and polycyclic aromatic hydrocarbons, which are able to absorb electromagnetic radiation (usually in the visible range of the spectrum) and eventually transfer that energy to molecular oxygen or the substrate of photooxygenation process. Many sensitizers, both naturally occurring and synthetic, rely on extensive aromatic systems to absorb light in the visible spectrum.[4] When sensitizers are excited by light, they reach a singlet state, 1Sens*. This singlet is then converted into a triplet state (which is more stable), 3Sens*, via intersystem crossing. The 3Sens* is what reacts with either the substrate or 3O2 in the three types of photooxygenation reactions.[6]

States of molecular oxygen
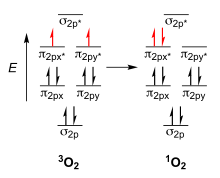
In classical Lewis structures, molecular oxygen, O2, is depicted as having a double bond between the two oxygen atoms. However, the molecular orbitals of O2 are actually more complex than Lewis structures seem to suggest. The highest occupied molecular orbital (HOMO) of O2 is a pair of degenerate antibonding π orbitals, π2px* and π2py*, which are both singly occupied by spin unpaired electrons.[4] These electrons are the cause of O2 being a triplet diradical in the ground state (indicated as 3O2).
While many stable molecules’ HOMOs consist of bonding molecular orbitals and therefore require a moderate energy jump from bonding to antibonding to reach their first excited state, the antibonding nature of molecular oxygen’s HOMO allows for a lower energy gap between its ground state and first excited state. This makes excitation of O2 a less energetically restrictive process. In the first excited state of O2, a 22 kcal/mol energy increase from the ground state, both electrons in the antibonding orbitals occupy a degenerate π* orbital, and oxygen is now in a singlet state (indicated as 1O2).[3] 1O2 is very reactive with a lifetime between 10-100µs.[4]
Types of photooxygenation
The three types of photooxygenation reactions are distinguished by the mechanisms that they proceed through, as they are capable of yielding different or similar products depending on environmental conditions. Type I and II reactions proceed through neutral intermediates, while type III reactions proceed through charged species. The absence or presence of 1O2 is what distinguishes type I and type II reactions, respectively.[1]
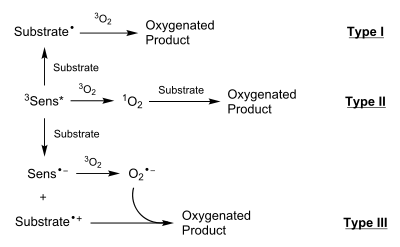
Type I
In type I reactions, the photoactivated 3Sens* interacts with the substrate to yield a radical substrate, usually through the homolytic bond breaking of a hydrogen bond on the substrate. This substrate radical then interacts with 3O2 (ground state) to yield a substrate-O2 radical. Such a radical is generally quenched by abstracting a hydrogen from another substrate molecule or from the solvent. This process allows for chain propagation of the reaction.
Example: Oxygen trapping of diradical intermediates
Type I photooxygenation reactions are frequently used in the process of forming and trapping diradical species. Mirbach et al. reported on one such reaction in which an azo compound is lysed via photolysis to form the diradical hydrocarbon and then trapped in a stepwise fashion by molecular oxygen:[7]
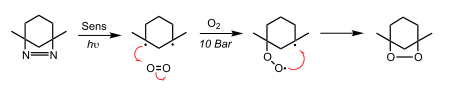
Type II
In type II reactions, the 3Sens* transfers its energy directly with 3O2 via a radiation-less transition to create 1O2. 1O2 then adds to the substrate in a variety of ways including: cycloadditions (most commonly [4+2]), addition to double bonds to yield 1,2-dioxetanes, and ene reactions with olefins.[2]
Example: precursor to prostaglandin synthesis
The [4+2] cycloaddition of singlet oxygen to cyclopentadiene to create cis-2-cyclopentene-1,4-diol is a common step involved in the synthesis of prostaglandins.[8] The initial addition singlet oxygen, through the concerted [4+2] cycloaddition, forms an unstable endoperoxide. Subsequent reduction of the peroxide bound produces the two alcohol groups.
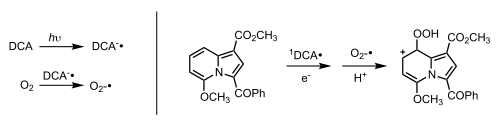
Type III
In type III reactions, there is an electron transfer that occurs between the 3Sens* and the substrate resulting in an anionic Sens and a cationic substrate. Another electron transfer then occurs where the anionic Sens transfers an electron to 3O2 to form the superoxide anion, O2−. This transfer returns the Sens to its ground state. The superoxide anion and cationic substrate then interact to form the oxygenated product.
Example: indolizine photooxygenation
Photooxygenation of indolizines (heterocyclic aromatic derivates of indole) has been investigated in both mechanistic and synthetic contexts. Rather than proceeding through a Type I or Type II photooxygenation mechanism, some investigators have chosen to use 9,10-dicyanoanthracene (DCA) as a photosensitzer, leading to the reaction of an indolizine derivative with the superoxide anion radical. Note that the reaction proceeds through an indolizine radical cation intermediate that has not been isolated (and thus is not depicted):[9]
Applications
Organic synthesis
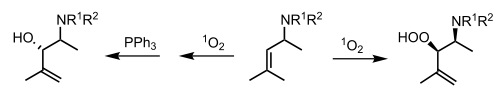
All 3 types of photooxygenation have been applied in the context of organic synthesis. In particular, type II photooxygenations have proven to be the most widely used (due to the low amount of energy required to generate singlet oxygen) and have been described as "one of the most powerful methods for the photochemical oxyfunctionalization of organic compounds."[10] These reactions can proceed in all common solvents and with a broad range of sensitizers.
Many of the applications of type II photooxygenations in organic synthesis come from Waldemar Adam's investigations into the ene-reaction of singlet oxygen with acyclic alkenes.[10] Through the cis effect and the presence of appropriate steering groups the reaction can even provide high regioselectively and diastereoselectivity - two valuable stereochemical controls.[11]
Photodynamic therapy
Photodynamic therapy (PDT) uses photooxygenation to destroy cancerous tissue.[12] A photosensitizer is injected into the tumor and then specific wavelengths of light are exposed to the tissue to excite the Sens. The excited Sens generally follows a type I or II photooxygenation mechanism to result in oxidative damage to cells. Extensive oxidative damage to tumor cells will kill tumor cells. Also oxidative damage to nearby blood vessels will cause local agglomeration and cut off nutrient supply to the tumor, thus starving the tumor.[13]
An important consideration when selecting the Sens to be used in PDT is the specific wavelength of light the Sens will absorb to reach an excited state. Since the maximum penetration of tissues is achieved around wavelengths of 800 nm, selecting Sens that absorb around this range is advantageous as it allows for PDT to be affective on tumors beneath the outer most layer of the dermis. The window of 800 nm light is most effective at penetrating tissues because at wavelengths shorter than 800 nm the light starts to be scattered by the macromolecules of cells and at wavelengths longer than 800 nm water molecules will begin to absorb the light and convert it into heat.[4]
References
- IUPAC (1997). A. D. McNaught and A. Wilkinson (ed.). Compendium of Chemical Terminology. Blackwell Scientific Publications, Oxford. doi:10.1351/goldbook. ISBN 978-0-9678550-9-7.
- M. R. Iesce; et al. (2005). "Photooxygenation of Heterocycles". Curr. Org. Chem. 9 (2): 109–139. doi:10.2174/1385272053369222.
- C.S. Foote (1968). "Mechanisms of Photosensitized Oxidation". Science. 162 (3857): 963–970. Bibcode:1968Sci...162..963F. doi:10.1126/science.162.3857.963.
- I. J. MacDonald, and T. J. Dougherty (2001). "Basic principles of photodynamic therapy". Journal of Porphyrins and Phthalocyanines. 5 (2): 105–129. doi:10.1002/jpp.328.
- Most of the recent (post-2000) literature includes the "Type III" classification; however, older articles only recognize Type I and Type II as named classes of photooxygenation reactions.
- C.S. Foote (1987). Type I and Type II Mechanisms of Photodynamic Action. ACS Symposium Series. 339. pp. 22–38. doi:10.1021/bk-1987-0339.ch002. ISBN 978-0-8412-1026-4.
- Mirbach, Marlis; M. Manfred; A. Saus (1982). "High-pressure photochemistry and ultraviolet spectroscopy in gas–liquid systems". Chemical Reviews. 82 (1): 59–76. doi:10.1021/cr00047a003.
- Stork, Gilbert; P. Sher; H. Chen (October 1986). "Radical Cyclization-Trapping in the Synthesis of Natural Products. A Simple, Stereocontrolled Route to Prostaglandin Fza". J. Am. Chem. Soc. 108 (20): 6384–6385. doi:10.1021/ja00280a043.
- Li, Yun; H. Hu; J. Ye; H. Fun; H. Hu; J. Xu (2004). "Reaction modes and mechanism in indolizine photooxygenation reactions". Journal of Organic Chemistry. 69 (7): 2332–2339. doi:10.1021/jo035070d.
- Rumbach, edited by Jochen Mattay and Axel G. Griesbeck in cooperation with Christian Stammel, Joachim Hirt and Thomas (1994). Jochen Mattay and Axel Griesbeck (ed.). Photochemical key steps in organic synthesis : an experimental course book. Weinheim: VCH. ISBN 978-3-527-29214-1.CS1 maint: extra text: authors list (link)
- Adam, Waldemar; W. Bruenker (1993). "Diastereoselective and regioselective photooxygenation of a chiral allylic amine and its acyl derivatives: stereochemical evidence for a steering effect by the amino group in the ene reaction of singlet oxygen". J. Am. Chem. Soc. 115 (7): 3008–3009. doi:10.1021/ja00060a072.
- Dougherty, Thomas (May 1987). "Photosensitizers: therapy and detection of malignant tumors". Photochemistry and Photobiology. 45 (445): 879–889. doi:10.1111/j.1751-1097.1987.tb07898.x.
- Chen, Qun; Z. Huang; H. Chen; H. Shapiro; J. Beckers; F. Hetzel (August 2002). "Improvement of Tumor Response by Manipulation of Tumor Oxygenation During Photodynamic Therapy". Photochemistry and Photobiology. 76 (2): 197–203. doi:10.1562/0031-8655(2002)0760197IOTRBM2.0.CO2.