Pathophysiology of acute respiratory distress syndrome
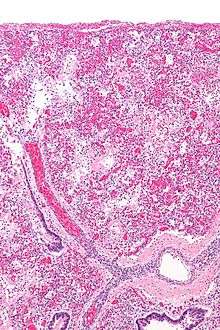
The pathophysiology of acute respiratory distress syndrome involves fluid accumulation in the lungs not explained by heart failure (noncardiogenic pulmonary edema). It is typically provoked by an acute injury to the lungs that results in flooding of the lungs' microscopic air sacs responsible for the exchange of gases such as oxygen and carbon dioxide with capillaries in the lungs.[1] Additional common findings in ARDS include partial collapse of the lungs (atelectasis) and low levels of oxygen in the blood (hypoxemia). The clinical syndrome is associated with pathological findings including pneumonia, eosinophilic pneumonia, cryptogenic organizing pneumonia, acute fibrinous organizing pneumonia, and diffuse alveolar damage (DAD). Of these, the pathology most commonly associated with ARDS is DAD, which is characterized by a diffuse inflammation of lung tissue. The triggering insult to the tissue usually results in an initial release of chemical signals and other inflammatory mediators secreted by local epithelial and endothelial cells.
| Pathophysiology of acute respiratory distress syndrome | |
|---|---|
 | |
| Biological system | respiratory system |
Neutrophils and some T-lymphocytes quickly migrate into the inflamed lung tissue and contribute in the amplification of the phenomenon. Typical histological presentation involves diffuse alveolar damage and hyaline membrane formation in alveolar walls. Although the triggering mechanisms are not completely understood, recent research has examined the role of inflammation and mechanical stress.
Inflammation
Inflammation, such as that caused by sepsis, causes endothelial cell dysfunction, fluid leakage from capillaries and impairs drainage of fluid from the lungs. Elevated inspired oxygen concentration often becomes necessary at this stage, and may facilitate a 'respiratory burst' in immune cells. In a secondary phase, endothelial cell dysfunction causes cells and inflammatory exudate to enter the alveoli. This pulmonary edema increases the thickness of the layer separating the blood in the capillary from the space in the air sacs, which increases the distance the oxygen must diffuse to reach the blood. This impairs gas exchange and leads to hypoxia, increased work of breathing, and eventually induces scarring of the air sacs of the lungs.
Fluid accumulation in the lungs and decreased surfactant production by type II pneumocytes may cause whole air sacs to collapse or to completely fill with fluid. This loss of aeration contributes further to the right-to-left shunt in ARDS. A traditional right-to-left shunt refers to blood passing from the right side of the heart to the left side without traveling to the capillaries of the lung for more oxygen (e.g., as seen in a patent foramen ovale). In ARDS, a lung right-to-left shunting occurs within the lungs since some blood from the right side of the heart will enter capillaries which cannot exchange gas with damaged air sacs that are full of fluid and debris from ARDS. As the alveoli contain progressively less gas, the blood flowing through the alveolar capillaries is progressively less oxygenated, resulting in massive shunting within the lung. The collapse of the air sacs and small airways interferes with the process of normal gas exchange. It is common to see patients with a PaO
2 of 60 mmHg (8.0 kPa) despite mechanical ventilation with 100% inspired oxygen.
The loss of aeration may follow different patterns depending upon the nature of the underlying disease and other factors. These are usually distributed to the lower lobes of the lungs, in their posterior segments, and they roughly correspond to the initial infected area. In sepsis or trauma-induced ARDS, infiltrates are usually more patchy and diffuse. The posterior and basal segments are always more affected, but the distribution is even less homogeneous. Loss of aeration also causes important changes in lung mechanical properties that are fundamental in the process of inflammation amplification and progression to ARDS in mechanically ventilated patients.
Mechanical stress
As the loss of aeration and the underlying disease progress, the end tidal volume grows to a level incompatible with life. Thus, mechanical ventilation is initiated to relieve muscles responsible for supporting breathing (respiratory muscles) of their work and to protect the affected person's airway. However, mechanical ventilation may constitute a risk factor for the development—or the worsening—of ARDS.[2] Aside from the infectious complications arising from invasive ventilation with endotracheal intubation, positive-pressure ventilation directly alters lung mechanics during ARDS. When these techniques are used the result is higher mortality through barotrauma.[2]
In 1998, Amato et al. published a paper showing substantial improvement in the outcome of patients ventilated with lower tidal volumes (Vt) (6 mL·kg−1).[2][3] This result was confirmed in a 2000 study sponsored by the NIH.[4] Both studies were widely criticized for several reasons, and the authors were not the first to experiment with lower-volume ventilation, but they increased the understanding of the relationship between mechanical ventilation and ARDS.
This form of stress is thought to be applied by the transpulmonary pressure (gradient) (Pl) generated by the ventilator or, better, its cyclical variations. The better outcome obtained in individuals ventilated with a lower Vt may be interpreted as a beneficial effect of the lower Pl.
The way Pl is applied on the alveolar surface determines the shear stress to which alveoli are exposed. ARDS is characterized by a usually heterogeneous reduction of the airspace, and thus by a tendency towards higher Pl at the same Vt, and towards higher stress on less diseased units. The heterogeneity of alveoli at different stages of disease is further increased by the gravitational gradient to which they are exposed and the different perfusion pressures at which blood flows through them.
The different mechanical properties of alveoli in ARDS may be interpreted as having varying time constants—the product of alveolar compliance × resistance. Slow alveoli are said to be "kept open" using PEEP, a feature of modern ventilators which maintains a positive airway pressure throughout the whole respiratory cycle. A higher mean pressure cycle-wide slows the collapse of diseased alveoli, but it has to be weighed against the corresponding elevation in Pl/plateau pressure. Newer ventilatory approaches attempt to maximize mean airway pressure for its ability to "recruit" collapsed alveoli while minimizing the shear stress caused by frequent openings and closings of aerated units.
Stress Index
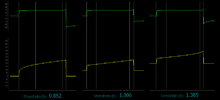
Mechanical ventilation can worsen the inflammatory response in people with ARDS by inducing hyperinflation of the alveoli and/or increased shear stress with frequent opening and closing of collapsible alveoli.[5] The stress index is measured during constant-flow volume assist-control mechanical ventilation without changing the baseline ventilatory pattern. Identifying the steadiest portion of the inspiratory flow (F) waveform fit the corresponding portion of the airway pressure (Paw) waveform in the following power equation:
Paw = a × tb + c where the coefficient b—the Stress Index—describes the shape of the curve. The Stress Index depicts a constant compliance if the value is around 1, an increasing compliance during the inspiration if the value is below 1, and a decreasing compliance if the value is above 1. Ranieri, Grasso, et al. set a strategy guided by the stress index with the following rules:
- Stress Index below 0.9, PEEP was increased
- Stress Index between 0.9 and 1.1, no change was made
- Stress Index above 1.1 PEEP was decreased.
Alveolar hyperinflation in patients with focal ARDS ventilated with the ARDSnet protocol is attenuated by a physiologic approach to PEEP setting based on the stress index measurement.[6]
Progression
If the underlying disease or injurious factor is not removed, the quantity of inflammatory mediators released by the lungs in ARDS may result in a systemic inflammatory response syndrome (SIRS) or sepsis if there is lung infection.[2] The evolution towards shock or multiple organ dysfunction syndrome follows paths analogous to the pathophysiology of sepsis. This leads to the impaired oxygenation, which is the central problem of ARDS, as well as to respiratory acidosis. Respiratory acidosis in ARDS is often caused by ventilation techniques such as permissive hypercapnia, which attempt to limit ventilator-induced lung injury in ARDS. The result is a critical illness in which the 'endothelial disease' of severe sepsis or SIRS is worsened by the lung dysfunction, which further impairs oxygen delivery to cells.
References
- Boyle, AJ; Mac Sweeney, R; McAuley, DF (August 2013). "Pharmacological treatments in ARDS; a state-of-the-art update". BMC Med. 11: 166. doi:10.1186/1741-7015-11-166. PMC 3765621. PMID 23957905.
- Irwin RS, Rippe JM (2003). Irwin and Rippe's Intensive Care Medicine (5th ed.). Lippincott Williams & Wilkins. ISBN 0-7817-3548-3.
- Amato M, Barbas C, Medeiros D, Magaldi R, Schettino G, Lorenzi-Filho G, Kairalla R, Deheinzelin D, Munoz C, Oliveira R, Takagaki T, Carvalho C (1998). "Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome". N Engl J Med. 338 (6): 347–54. doi:10.1056/NEJM199802053380602. PMID 9449727.
- MacIntyre N (2000). "Mechanical ventilation strategies for lung protection". Semin Respir Crit Care Med. 21 (3): 215–22. doi:10.1055/s-2000-9850. PMID 16088734.
- Slutsky AS (May 2005). "Ventilator-induced lung injury: from barotrauma to biotrauma" (PDF). Respir Care. 50 (5): 646–59. PMID 15912625.
- Grasso S, Stripoli T, De Michele M, et al. (October 2007). "ARDSnet ventilatory protocol and alveolar hyperinflation: role of positive end-expiratory pressure". Am. J. Respir. Crit. Care Med. 176 (8): 761–7. doi:10.1164/rccm.200702-193OC. PMID 17656676.