Oxoammonium-catalyzed oxidation
Oxoammonium-catalyzed oxidation reactions involve the conversion of organic substrates to more highly oxidized materials through the action of an N-oxoammonium species. Nitroxides may also be used in catalytic amounts in the presence of a stoichiometric amount of a terminal oxidant.[1] Nitroxide radical species used are either 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) or derivatives thereof.
(1)

Mechanism and stereochemistry
One-electron oxidation of the nitroxide produces a highly electrophilic oxoammonium species, which serves as the active oxidizing agent.[2] The nitroxide can be used as a catalyst in conjunction with cheaper stoichiometric oxidants such as sodium hypochlorite[3] or bis(acetoxy)iodobenzene (BAIB).[4]
Under neutral or slightly acidic conditions (in the presence of silica gel, for instance), oxidation occurs by an initial hydrogen bond between the hydroxyl group and the oxoammonium nitrogen, followed by concerted proton transfer and hydride abstraction.[5] The need for hydrogen bonding is supported by the low reactivity of β-alkoxy and β-amino alcohols, which exhibit competitive intramolecular hydrogen bonding. The mechanism of oxidation under weakly basic (pyridine) conditions is similar, except that pyridine neutralizes the hydroxyammonium species, and this intermediate "comproportionates" with oxoammonium salt to give nitroxide radicals and pyridinium salts (see equation (3) below). Because this reaction consumes base and active oxidant, two equivalents of base and oxidant are necessary under weakly basic conditions. A unified mechanism under neutral and basic conditions in presented in a recent article.[6] The authors present a comprehensive analysis of a number of oxoammonium salt mediated oxidations.
(2)

Under strongly basic conditions, the deprotonated substrate reacts with the N-oxyammonium species. Attack of the substrate alkoxide on either nitrogen or oxygen may occur, although the former is believed to operate on the basis of on observations of oxidations of N-alkoxy amines (which, presumably, proceed via intermediate 1).[7] Comproportionation of the reduced product (a hydroxylamine) with the oxoammonium ion competes with oxidation; thus, an excess of the oxidizing agent is often required.
(3)

Nitroxide-catalyzed oxidations involve N-oxoammonium intermediates as the active oxidizing agent. The mechanism of oxidation of the nitroxide radical depends on the terminal oxidant employed. Two-electron oxidants, such as NaOCl, are able to directly convert nitroxides into oxoammoniums.
(4)

One-electron oxidants, such as copper(II), operate via a more complex mechanism involving dioxygen as the terminal oxidant.[8] Copper(II) oxidizes four equivalents of nitroxide to oxoammonium, two equivalents of which (blue) react with alcohols to form carbonyl compounds. The other two equivalents of oxoammonium (red) undergo comproportionation to re-form nitroxy radicals (pink). Finally, dioxygen re-oxidizes four equivalents of copper(I) back to copper(II). Overall, a single molecule of dioxygen mediates the oxidation of two equivalents of alcohol, with the formation of two equivalents of water.
(5)

Stereoselective variants
Enantioselective oxidations are typically either kinetic resolutions of chiral alcohols or desymmetrization reactions. These oxidations may be facilitated through the use of chiral nitroxide radicals in the catalytic mode. A good example is provided by the kinetic resolution of racemic 1-phenylethanol.[9] Oxidative desymmetrization processes employing oxoammonium oxidants, on the other hand, are relatively rare.[10]
(6)

Scope
Oxidations using oxoammonium salts may be carried out either in the stoichiometric or catalytic mode under acidic or basic conditions. This section describes the most commonly used conditions for the stoichiometric and catalytic oxidation of alcohols to carbonyl compounds with oxoammonium salts. Although a wide variety of alcohols may be oxidized using TEMPO, competitive oxidation of more electron-rich functionality sometimes takes place. In addition, the site selectivity of oxidation of polyols may differ depending on the conditions used.
Stoichiometric oxidations
Under mildly acidic or neutral conditions, oxoammonium salts such as Bobbitt's salt oxidize allylic, benzylic,[11] propargylic,[12] or aliphatic alcohols to the corresponding aldehydes or ketones. Secondary alcohols react faster than primary ones, although selectivity is low. A convenient experimental protocol allows for recycling of the oxoammonium salt.[12]
(7)
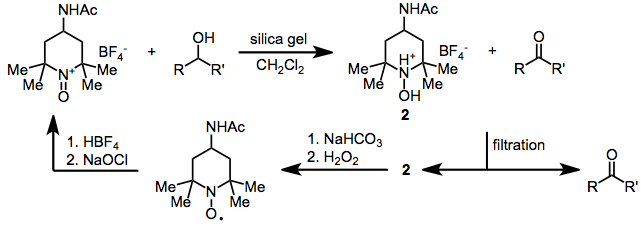
Amines, benzylic ethers, and alkenes are oxidized more rapidly than unactivated alcohols; thus, selective stoichiometric oxidation of unactivated alcohols in the presence of these functional groups is not possible.[13] Alcohols with β-nitrogen or β-oxygen substituents react sluggishly under acidic conditions.[12] Allylic and benzylic alcohols can be selectively oxidized under these conditions[13]
(8)

Under basic conditions, two equivalents of oxidant are needed because of competitive comproportionation between reduced nitroxide and unreacted oxoammonium (see equation (3) above). Pyridine is usually employed as the base. These are the most common conditions for nitroxide oxidations in the stoichiometric mode.
(9)

Catalytic oxidations
Catalytic oxoammonium oxidation may be facilitated using sodium hypochlorite as the terminal oxidant. The pH must be maintained below 10 using a buffer for the reaction to proceed. The active oxidizing agent of nitroxide is hypobromite anion; hence, potassium bromide is used as an additive.[3] No epimerization of α-stereogenic centers in carbonyl-containing products takes place.
(10)

The use of chlorites as terminal oxidants in conjunction with both hypochlorites and TEMPO gives carboxylic acids without chlorination side products.[14] The reaction is usually carried out in two steps in the same pot: partial oxidation is effected with TEMPO and hypochlorite, then chlorite is added to complete the oxidation. Only primary alcohol oxidation is observed. In conjunction with Sharpless dihydroxylation, this method can be used to generate enantiopure α-hydroxy acids.[15]
(11)

A significant limitation of both of the above methods is incompatibility with free amine or alkene functionality, both of which undergo competitive oxidation. The use of bis(acetoxy)iodobenzene (BAIB) as the terminal oxidant avoids this problem. BAIB is unable to oxidize the nitroxide radical directly, and initial formation of oxoammonium is believed to be due to acid-catalyzed disproportionation. BAIB may then oxidize the resulting hydroxylamine to an oxoammonium salt. Although the reaction is conducted under acidic conditions (acetic acid is a byproduct, and is often added to facilitate disproportionation), selectivity for primary alcohol oxidation is substantial.[4] Base-sensitive functional groups, such as epoxides, are tolerated under these conditions.[16]
(12)

Other two-electron terminal oxidants used with TEMPO include mCPBA (secondary oxidation is favored, although side reactions may occur),[17] N-chlorosuccinimide,[18] and Oxone.[19]
Copper(II), both as the free chloride salt and as a complex with bidentate ligands, oxidizes TEMPO to its oxoammonium salt. In these reactions, air serves as the terminal oxidant.[20] It is unclear whether air oxidizes copper(I) to copper(II), or whether alcohol oxidation is partially mediated by copper and air oxidizes the resulting hydroxylamine back to the oxoammonium salt. The former occurs during the Wacker process, but the latter explains why copper complexes and a few other metal complexes are able to oxidize alcohols in conjunction with TEMPO.
(13)

Comparison with related methods
Activated manganese dioxide, which oxidizes allylic and benzylic alcohols, is cheaper than TEMPO and operationally simple to use.[21] Chromium-based reagents such as pyridinium chlorochromate can also be used to convert alcohols to carbonyl compounds; although the stoichiometric generation of chromium wastes is a disadvantage.[22] Oxidations employing dimethyl sulfoxide, such as the Swern and Moffatt reactions, involve no heavy metals and oxidize a wide variety of substrates.[23] Oxoammonium oxidations are preferred to DMSO methods for reactions of diols and acetylenic alcohols. Dess-Martin periodinane is a highly selective, mild oxidant of alcohols, whose primary disadvantages are difficulties with preparation and safety.[24]
References
- Bobbitt, J. M.; Bruckner, C.; Merbouh, N. Org. React. 2009, 74, 103. doi:10.1002/0471264180.or074.02
- Merbouh, N.; Bobbitt, J. M.; Brückner, C. J. Org. Chem. 2004, 69, 5116.
- Sheldon, R. A.; Arends, I. W. C. E.; ten Brink, G. J.; Dijksman, A. Acc. Chem. Res. 2002, 35, 774. doi:10.1021/ar010075n
- De Mico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. J. Org. Chem. 1997, 62, 6974.
- Bailey, W. F.; Bobbitt, J. M.; Wiberg, K. B. J. Org. Chem. 2007, 72, 4504.
- Hamlin, T. A.; Kelly, C. B.; Ovian, J. M.; Wiles, R. J.; Tilley, L. J.; Leadbeater, N. E. J. Org. Chem. 2015, 80, 8150.
- Semmelhack, M. F.; Schmid, C. R.; Cortés, D. A. Tetrahedron Lett. 1986, 27, 1119.
- Semmelhack, M. F.; Schmid, C. R.; Cortés, D. A.; Chou, C. S. J. Am. Chem. Soc. 1984, 106, 3374.
- Rychnovsky, S. D.; McLernon, T. L.; Rajapakse, H. J. Org. Chem. 1996, 61, 1194.
- Tanaka, H.; Kawakami, Y.; Goto, K.; Kuroboshi, M. Tetrahedron Lett. 2001, 42, 445.
- Miyazawa, T.; Endo, T.; Shiihashi, S.; Okawara, M. J. Org. Chem. 1985, 50, 1332.
- Bobbitt, J. M. J. Org. Chem. 1998, 63, 9367.
- Bobbitt, J. M.; Merbouh, N. Org. Synth. 2005, 82, 80.>
- Song, Z. J.; Zhao, M.; Desmond, R.; Devine, P.; Tschaen, D. M.; Tillyer, R.; Frey, L.; Heid, R.; Xu, F.; Foster, B.; Li, J.; Reamer, R.; Volante, R.; Grabowski, E. J. J.; Dolling, U. H.; Reider, P. J.; Okada, S.; Kato, Y.; Mano, E. J. Org. Chem. 1999, 64, 9658.
- Sharpless, K. B.; Amberg, W.; Bennani, Y. L.; Crispino, G. A.; Hartung, J.; Jeong, K. S.; Kwong, H. L.; Morikawa, K.; Wang, Z. M.; Xu, D.; Zhang, X. L. J. Org. Chem. 1992, 57, 2768.
- De Mico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. J. Org. Chem. 1997, 62, 6974.
- Ganem, B. J. Org. Chem. 1975, 40, 1998.
- Einhorn, J.; Einhorn, C.; Ratajczak, F.; Pierre, J.-L. J. Org. Chem. 1996, 61, 7452.
- Bolm, C.; Magnus, A. S.; Hildebrand, J. P. Org. Lett. 2000, 2, 1173.
- Sheldon, R. A.; Arends, I. W. C. E. Adv. Synth. Catal. 2004, 346, 1051.
- Taylor, R. J. K.; Reid, M.; Foot, J.; Raw, S. A. Acc. Chem. Res. 2005, 38, 851.
- Luzzio, F. A. Org. React. 1998, 53, 1.
- Tidwell, T. T. Org. React. 1990, 39, 297.
- Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277.