Machine learning in bioinformatics
Machine learning, a subfield of computer science involving the development of algorithms that learn how to make predictions based on data, has a number of emerging applications in the field of bioinformatics. Bioinformatics deals with computational and mathematical approaches for understanding and processing biological data.[1]
Prior to the emergence of machine learning algorithms, bioinformatics algorithms had to be explicitly programmed by hand which, for problems such as protein structure prediction, proves extremely difficult.[2] Machine learning techniques such as deep learning enable the algorithm to make use of automatic feature learning which means that based on the dataset alone, the algorithm can learn how to combine multiple features of the input data into a more abstract set of features from which to conduct further learning. This multi-layered approach to learning patterns in the input data allows such systems to make quite complex predictions when trained on large datasets. In recent years, the size and number of available biological datasets have skyrocketed, enabling bioinformatics researchers to make use of these machine learning systems.[3] Machine learning has been applied to six biological domains: genomics, proteomics, microarrays, systems biology, evolution, and text mining.[3]
Applications
Genomics
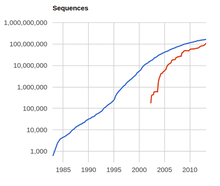
Genomics involves the study of the genome, the complete DNA sequence, of organisms. While genomic sequence data has historically been sparse due to the technical difficulty in sequencing a piece of DNA, the number of available sequences is growing exponentially.[4] However, while raw data is becoming increasingly available and accessible, the biological interpretation of this data is occurring at a much slower pace.[5] Therefore, there is an increasing need for the development of machine learning systems that can automatically determine the location of protein-encoding genes within a given DNA sequence.[5] This is a problem in computational biology known as gene prediction.
Gene prediction is commonly performed through a combination of what are known as extrinsic and intrinsic searches.[5] For the extrinsic search, the input DNA sequence is run through a large database of sequences whose genes have been previously discovered and their locations annotated. A number of the sequence's genes can be identified by determining which strings of bases within the sequence are homologous to known gene sequences. However, given the limitation in size of the database of known and annotated gene sequences, not all the genes in a given input sequence can be identified through homology alone. Therefore, an intrinsic search is needed where a gene prediction program attempts to identify the remaining genes from the DNA sequence alone.[5]
Machine learning has also been used for the problem of multiple sequence alignment which involves aligning many DNA or amino acid sequences in order to determine regions of similarity that could indicate a shared evolutionary history.[3] It can also be used to detect and visualize genome rearrangements.[6]
Proteomics

Proteins, strings of amino acids, gain much of their function from protein folding in which they conform into a three-dimensional structure. This structure is composed of a number of layers of folding, including the primary structure (i.e. the flat string of amino acids), the secondary structure (alpha helices and beta sheets), the tertiary structure, and the quartenary structure.
Protein secondary structure prediction is a main focus of this subfield as the further protein foldings (tertiary and quartenary structures) are determined based on the secondary structure.[2] Solving the true structure of a protein is an incredibly expensive and time-intensive process, furthering the need for systems that can accurately predict the structure of a protein by analyzing the amino acid sequence directly.[2][3] Prior to machine learning, researchers needed to conduct this prediction manually. This trend began in 1951 when Pauling and Corey released their work on predicting the hydrogen bond configurations of a protein from a polypeptide chain.[7] Today, through the use of automatic feature learning, the best machine learning techniques are able to achieve an accuracy of 82-84%.[2][8] The current state-of-the-art in secondary structure prediction uses a system called DeepCNF (deep convolutional neural fields) which relies on the machine learning model of artificial neural networks to achieve an accuracy of approximately 84% when tasked to classify the amino acids of a protein sequence into one of three structural classes (helix, sheet, or coil).[8] The theoretical limit for three-state protein secondary structure is 88–90%.[2]
Machine learning has also been applied to proteomics problems such as protein side-chain prediction, protein loop modeling, and protein contact map prediction.[3]
Microarrays
Microarrays, a type of lab-on-a-chip, are used for automatically collecting data about large amounts of biological material. Machine learning can aid in the analysis of this data, and it has been applied to expression pattern identification, classification, and genetic network induction.[3]
This technology is especially useful for monitoring the expression of genes within a genome, aiding in diagnosing different types of cancer based on which genes are expressed.[9] One of the main problems in this field is identifying which genes are expressed based on the collected data.[3] In addition, due to the huge number of genes on which data is collected by the microarray, there is a large amount of irrelevant data to the task of expressed gene identification, further complicating this problem. Machine learning presents a potential solution to this problem as various classification methods can be used to perform this identification. The most commonly used methods are radial basis function networks, deep learning, Bayesian classification, decision trees, and random forest.[9]
Systems biology
Systems biology focuses on the study of the emergent behaviors from complex interactions of simple biological components in a system. Such components can include molecules such as DNA, RNA, proteins, and metabolites.[10]
Machine learning has been used to aid in the modelling of these complex interactions in biological systems in domains such as genetic networks, signal transduction networks, and metabolic pathways.[3] Probabilistic graphical models, a machine learning technique for determining the structure between different variables, are one of the most commonly used methods for modeling genetic networks.[3] In addition, machine learning has been applied to systems biology problems such as identifying transcription factor binding sites using a technique known as Markov chain optimization.[3] Genetic algorithms, machine learning techniques which are based on the natural process of evolution, have been used to model genetic networks and regulatory structures.[3]
Other systems biology applications of machine learning include the task of enzyme function prediction, high throughput microarray data analysis, analysis of genome-wide association studies to better understand markers of disease, protein function prediction.[11]
Stroke Diagnosis
Machine learning methods for analysis of neuroimaging data are used to help diagnose stroke. Three-dimensional CNN and SVM methods are often used. [12]
Text mining
The increase in available biological publications led to the issue of the increase in difficulty in searching through and compiling all the relevant available information on a given topic across all sources. This task is known as knowledge extraction. This is necessary for biological data collection which can then in turn be fed into machine learning algorithms to generate new biological knowledge.[3][13] Machine learning can be used for this knowledge extraction task using techniques such as natural language processing to extract the useful information from human-generated reports in a database. Text Nailing, an alternative approach to machine learning, capable of extracting features from clinical narrative notes was introduced in 2017.
This technique has been applied to the search for novel drug targets, as this task requires the examination of information stored in biological databases and journals.[13] Annotations of proteins in protein databases often do not reflect the complete known set of knowledge of each protein, so additional information must be extracted from biomedical literature. Machine learning has been applied to automatic annotation of the function of genes and proteins, determination of the subcellular localization of a protein, analysis of DNA-expression arrays, large-scale protein interaction analysis, and molecule interaction analysis.[13]
Another application of text mining is the detection and visualization of distinct DNA regions given sufficient reference data.[14]
References
- Chicco D (December 2017). "Ten quick tips for machine learning in computational biology". BioData Mining. 10 (35): 35. doi:10.1186/s13040-017-0155-3. PMC 5721660. PMID 29234465.
- Yang, Yuedong; Gao, Jianzhao; Wang, Jihua; Heffernan, Rhys; Hanson, Jack; Paliwal, Kuldip; Zhou, Yaoqi (May 2018). "Sixty-five years of the long march in protein secondary structure prediction: the final stretch?". Briefings in Bioinformatics. 19 (3): 482–494. doi:10.1093/bib/bbw129. PMC 5952956. PMID 28040746.
- Larrañaga, Pedro; Calvo, Borja; Santana, Roberto; Bielza, Concha; Galdiano, Josu; Inza, Iñaki; Lozano, José A.; Armañanzas, Rubén; Santafé, Guzmán (March 2006). "Machine learning in bioinformatics". Briefings in Bioinformatics. 7 (1): 86–112. doi:10.1093/bib/bbk007. PMID 16761367.
- "GenBank and WGS Statistics". www.ncbi.nlm.nih.gov. Retrieved May 6, 2017.
- Mathé, Catherine; Sagot, Marie-France; Schiex, Thomas; Rouzé, Pierre (October 1, 2002). "Current methods of gene prediction, their strengths and weaknesses". Nucleic Acids Research. 30 (19): 4103–4117. doi:10.1093/nar/gkf543. ISSN 1362-4962. PMC 140543. PMID 12364589.
- Pratas, D; Silva, R; Pinho, A; Ferreira, P (May 18, 2015). "An alignment-free method to find and visualise rearrangements between pairs of DNA sequences". Scientific Reports. 5 (10203): 10203. Bibcode:2015NatSR...510203P. doi:10.1038/srep10203. PMC 4434998. PMID 25984837.
- Pauling, L.; Corey, R. B.; Branson, H. R. (April 1, 1951). "The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain". Proceedings of the National Academy of Sciences of the United States of America. 37 (4): 205–211. Bibcode:1951PNAS...37..205P. doi:10.1073/pnas.37.4.205. ISSN 0027-8424. PMC 1063337. PMID 14816373.
- Wang, Sheng; Peng, Jian; Ma, Jianzhu; Xu, Jinbo (December 1, 2015). "Protein secondary structure prediction using deep convolutional neural fields". Scientific Reports. 6: 18962. arXiv:1512.00843. Bibcode:2016NatSR...618962W. doi:10.1038/srep18962. PMC 4707437. PMID 26752681.
- Pirooznia, Mehdi; Yang, Jack Y.; Yang, Mary Qu; Deng, Youping (2008). "A comparative study of different machine learning methods on microarray gene expression data". BMC Genomics. 9 (1): S13. doi:10.1186/1471-2164-9-S1-S13. ISSN 1471-2164. PMC 2386055. PMID 18366602.
- "Machine Learning in Molecular Systems Biology". Frontiers. Retrieved June 9, 2017.
- d'Alché-Buc, Florence; Wehenkel, Louis (2008). "Machine Learning in Systems Biology". BMC Proceedings. 2 (4): S1. doi:10.1186/1753-6561-2-S4-S1. ISSN 1753-6561. PMC 2654969. PMID 19091048.
- Jiang, Fei (2017). "Artificial intelligence in healthcare: past, present and future" (PDF). BMJ Journals Stroke and Vascular Neurology. 2 (4): 230–243. doi:10.1136/svn-2017-000101. PMC 5829945. PMID 29507784. Retrieved January 23, 2019.
- Krallinger, Martin; Erhardt, Ramon Alonso-Allende; Valencia, Alfonso (March 15, 2005). "Text-mining approaches in molecular biology and biomedicine". Drug Discovery Today. 10 (6): 439–445. doi:10.1016/S1359-6446(05)03376-3. PMID 15808823.
- Pratas, D; Hosseini, M; Silva, R; Pinho, A; Ferreira, P (June 20–23, 2017). Visualization of Distinct DNA Regions of the Modern Human Relatively to a Neanderthal Genome. Iberian Conference on Pattern Recognition and Image Analysis. Springer. Lecture Notes in Computer Science. 10255. pp. 235–242. doi:10.1007/978-3-319-58838-4_26. ISBN 978-3-319-58837-7.