I-TASSER
I-TASSER (Iterative Threading ASSEmbly Refinement) is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences.[1] It detects structure templates from the Protein Data Bank by a technique called fold recognition (or threading). The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
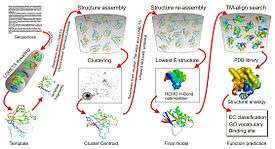 I-TASSER pipeline for protein structure and function prediction. | |
| Developer(s) | Yang Zhang Lab |
|---|---|
| Website | zhanglab |
I-TASSER has been extended for structure-based protein function predictions, which provides annotations on ligand binding site, gene ontology and enzyme commission by structurally matching structural models of the target protein to the known proteins in protein function databases.[2][3] It has an on-line server built in the Yang Zhang Lab at the University of Michigan, Ann Arbor, allowing users to submit sequences and obtain structure and function predictions. A standalone package of I-TASSER is available for download at the I-TASSER website.
Ranking in CASP
I-TASSER (as 'Zhang-Server') has been consistently ranked as the top method in CASP, a community-wide experiment to benchmark the best structure prediction methods in the field of protein folding and protein structure prediction. CASP takes place every two years since 1994.[4]
- No 1 in CASP7 (2006) [5]
- No 1 in CASP8 (2008): Official ranking of CASP8 (164 targets)
- No 2 in CASP9 (2010): Official ranking of CASP9 (147 targets)
- No 1 in CASP10 (2012): Official ranking of CASP10 (127 targets)
- No 1 in CASP11 (2014): Official ranking of CASP11 (126 targets)
- No 1 in CASP12 (2016): Official ranking of CASP12 (96 targets)
Method and pipeline
I-TASSER is a template-based method for protein structure and function prediction.[1] The pipeline consists of six consecutive steps:
- 1, Secondary structure prediction by PSSpred
- 2, Template detection by LOMETS[6]
- 3, Fragment structure assembly using replica-exchange Monte Carlo simulation[7]
- 4, Model selection by clustering structure decoys using SPICKER[8]
- 5, Atomic-level structure refinement by fragment-guided molecular dynamics simulation (FG-MD)[9] or ModRefiner[10]
- 6, Structure-based biology function annotation by COACH[11]
On-line Server
The I-TASSER server allows users to generate automatically protein structure and function predictions.
- Input
- Mandatory:
- Amino acid sequence with length from 10 to 1,500 residues
- Optional (user can provide optionally restraints and templates to assist I-TASSER modeling):
- Contact restraints
- Distance maps
- Inclusion of special templates
- Exclusion of special templates
- Secondary structures
- Mandatory:
- Output
- Structure prediction:
- Secondary structure prediction
- Solvent accessibility prediction
- Top 10 threading alignment from LOMETS
- Top 5 full-length atomic models (ranked based on cluster density)
- Top 10 proteins in PDB which are structurally closest to the predicted models
- Estimated accuracy of the predicted models (including a confidence score of all models, predicted TM-score and RMSD for the first model, and per-residue error of all models)
- B-factor estimation
- Function prediction:
- Enzyme Classification (EC) and the confidence score
- Gene Ontology (GO) terms and the confidence score
- Ligand-binding sites and the confidence score
- An image of the predicted ligand-binding sites
- Structure prediction:
Standalone Suite
The I-TASSER Suite is a downloadable package of standalone computer programs, developed by the Yang Zhang Lab for protein structure prediction and refinement, and structure-based protein function annotations.[12] Through the I-TASSER License, researchers have access to the following standalone programs:
- I-TASSER: A standalone I-TASSER package for protein 3D structure prediction and refinement.
- COACH: A function annotation program based on COFACTOR, TM-SITE and S-SITE.
- COFACTOR: A program for ligand-binding site, EC number & GO term prediction.
- TM-SITE: A structure-based approach for ligand-binding site prediction.
- S-SITE: A sequence-based approach for ligand-binding site prediction.
- LOMETS: A set of locally installed threading programs for meta-server protein fold-recognition.
- MUSTER: A threading program to identify templates from a non-redundant protein structure library.
- SPICKER: A clustering program to identify near-native protein model from structure decoys.
- HAAD: A program for quickly adding hydrogen atoms to protein heavy-atom structures.
- EDTSurf: A program to construct triangulated surfaces of protein molecules.
- ModRefiner: A program to construct and refine atomic-level protein models from C-alpha traces.
- NW-align: A robust program for protein sequence-to-sequence alignments by Needleman-Wunsch algorithm.
- PSSpred: A highly accurate program for protein secondary structure prediction.
- Library: I-TASSER structural and functional template library weekly updated and freely accessible to the I-TASSER users.
Help documents
- Instruction on how to download and install the I-TASSER Suite can be found at README.txt.
References
- Roy A, Kucukural A, Zhang Y (2010). "I-TASSER: a unified platform for automated protein structure and function prediction". Nature Protocols. 5 (4): 725–738. doi:10.1038/nprot.2010.5. PMC 2849174. PMID 20360767.
- Roy A, Yang J, Zhang Y (2012). "COFACTOR: An accurate comparative algorithm for structure-based protein function annotation". Nucleic Acids Research. 40 (Web Server issue): W471–W477. doi:10.1093/nar/gks372. PMC 3394312. PMID 22570420.
- Zhang C, Freddolino PL, Zhang Y (2017). "COFACTOR: improved protein function prediction by combining structure, sequence and protein-protein interaction information". Nucleic Acids Research. 45 (W1): W291–W299. doi:10.1093/nar/gkx366. PMC 5793808. PMID 28472402.
- Moult, J; et al. (1995). "A large-scale experiment to assess protein structure prediction methods" (PDF). Proteins. 23 (3): ii–iv. doi:10.1002/prot.340230303. PMID 8710822.
- Battey, JN; et al. (2007). "Automated server predictions in CASP7". Proteins. 69 (Suppl 8): 68–82. doi:10.1002/prot.21761. PMID 17894354.
- Wu S, Zhang Y (2007). "LOMETS: A local meta-threading-server for protein structure prediction". Nucleic Acids Research. 35 (10): 3375–3382. doi:10.1093/nar/gkm251. PMC 1904280. PMID 17478507.
- Swendsen RH, Wang JS (1986). "Replica Monte Carlo simulation of spin glasses". Physical Review Letters. 57 (21): 2607–2609. doi:10.1103/physrevlett.57.2607. PMID 10033814.
- Zhang Y, Skolnick J (2004). "SPICKER: A Clustering Approach to Identify Near-Native Protein Folds". Journal of Computational Chemistry. 25 (6): 865–871. doi:10.1002/jcc.20011. PMID 15011258.
- Zhang J, Liang Y, Zhang Y (2011). "Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling". Structure. 19 (12): 1784–1795. doi:10.1016/j.str.2011.09.022. PMC 3240822. PMID 22153501.
- Xu D, Zhang Y (2011). "Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-step Atomic-level Energy Minimization". Biophysical Journal. 101 (10): 2525–2534. doi:10.1016/j.bpj.2011.10.024. PMC 3218324. PMID 22098752.
- Yang J, Roy A, Zhang Y (2013). "Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment". Bioinformatics. 29 (20): 2588–2595. doi:10.1093/bioinformatics/btt447. PMC 3789548. PMID 23975762.
- Yang J, Roy A, Zhang Y (2015). "The I-TASSER Suite: Protein structure and function prediction". Nature Methods. 12 (1): 7–8. doi:10.1038/nmeth.3213. PMC 4428668. PMID 25549265.