Hyperphenylalaninemia
Hyperphenylalaninemia is a medical condition characterized by mildly or strongly elevated concentrations of the amino acid phenylalanine in the blood. Phenylketonuria (PKU) can result in severe hyperphenylalaninemia.[1] Phenylalanine concentrations ([phe]) are routinely screened in newborns by the neonatal heel prick (Guthrie test), which takes a few drops of blood from the heel of the infant. Standard [phe] concentrations in unaffected persons are about 60µM: [phe] concentrations in persons with untreated phenylketonuria may be many times that (600µM to 2400µM), which indicate that the child is at risk for severe intellectual disability. Phenylketonuria is classed as an autosomal recessive condition: in heterozygous form, [phe] shows a moderate elevation, perhaps two-fold over that of unaffected homozygotes, which is classified as hyperphenylalaninemia (hyper- + phenylalanine + -emia = high [phe] in blood).
| Hyperphenylalaninemia | |
|---|---|
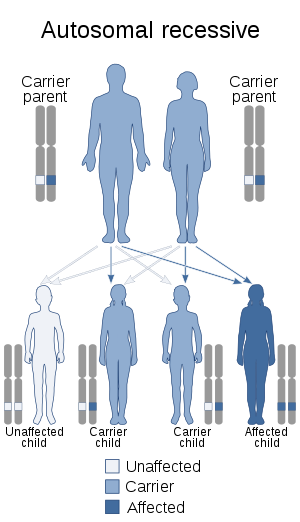 | |
| Hyperphenylalaninemia can be inherited in an autosomal recessive manner. | |
| Specialty | Endocrinology |
Symptoms
The coloration of the skin, hair, and eyes is different in children with PKU. This is caused by low levels of tyrosine, whose metabolic pathway is blocked by deficiency of PAH. Another skin alteration that might occur is the presence of irritation or dermatitis. The child's behaviour may be influenced as well due to augmented levels of phenethylamine which in turn affects levels of other amines in the brain. Psychomotor function may be affected and observed to worsen progressively.
Cause
People with the genotype for PKU are unaffected in utero, because maternal circulation prevents buildup of [phe]. After birth, PKU in newborns is treated by a special diet with highly restricted phenylalanine content. Persons with genetic predisposition to PKU have normal mental development on this diet. Previously, it was thought safe to withdraw from the diet in the late teens or early twenties, after the central nervous system was fully developed; recent studies suggest some degree of relapse, and a continued phenylalanine-restricted diet is now recommended.
PKU or hyperphenylalaninemia may also occur in persons without the PKU genotype. If the mother has the PKU genotype but has been treated so as to be asymptomatic, high levels of [phe] in the maternal blood circulation may affect the non-PKU fetus during gestation. Mothers successfully treated for PKU are advised to return to the [phe]-restricted diet during pregnancy.
A small subset of patients with hyperphenylalaninemia shows an appropriate reduction in plasma phenylalanine levels with dietary restriction of this amino acid; however, these patients still develop progressive neurologic symptoms and seizures and usually die within the first 2 years of life ("malignant" hyperphenylalaninemia). These infants exhibit normal phenylalanine hydroxylase (PAH) enzymatic activity but have a deficiency in dihydropteridine reductase (DHPR), an enzyme required for the regeneration of tetrahydrobiopterin (THB or BH4), a cofactor of PAH.
Less frequently, DHPR activity is normal but a defect in the biosynthesis of THB exists. In either case, dietary therapy corrects the hyperphenylalaninemia. However, THB is also a cofactor for two other hydroxylation reactions required in the syntheses of neurotransmitters in the brain: the hydroxylation of tryptophan to 5-hydroxytryptophan and of tyrosine to L-dopa. It has been suggested that the resulting deficit in the CNS neurotransmitter activity is, at least in part, responsible for the neurologic manifestations and eventual death of these patients.[2] Hyperphenylalaninemia most is commonly diagnosed by newborn screening and must be distinguished from classic PKU by confirmatory testing at an experienced center. Some cases in adult women have been detected using maternal screening programs or following birth of children with birth defects. Elevated phenylalanine levels are associated with neuropsychological effects.
Treatment
Maintain plasma phenylalanine values in therapeutic range of 120 to 360 mM using a diet that restricts phenylalanine but otherwise nutritionally complete. Treatment for life is recommended to reduce the risk of long term neuropsychiatric problems and reduce the risk of maternal PKU syndrome.
Outcome
With treatment the outcome is excellent. Most infants with classic PKU who are treated within the first 10 days of life achieve normal intelligence. However learning problems are more frequent than in unaffected peers.
References
- "OMIM Entry # 261600 - Phenylketonuria; PKU". omim.org. Archived from the original on 2014-05-29. Retrieved 2016-06-03.
- "Basic Medical Biochemistry", Fourth edition
External links
| Classification | |
|---|---|
| External resources |