Hybrid genome assembly
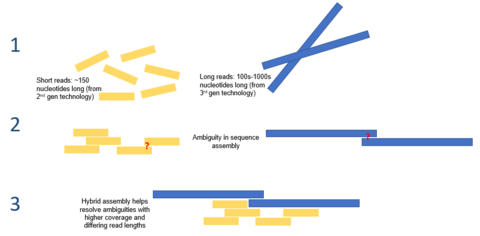
In bioinformatics, hybrid genome assembly refers to utilizing various sequencing technologies to achieve the task of assembling a genome from fragmented, sequenced DNA resulting from shotgun sequencing. Genome assembly presents one of the most challenging tasks in genome sequencing as most modern DNA sequencing technologies can only produce reads that are, on average, 25-300 base pairs in length.[1] This is orders of magnitude smaller than the average size of a genome (the genome of the octoploid plant Paris japonica is 149 billion base pairs[2]). This assembly is computationally difficult and has some inherent challenges, one of these challenges being that genomes often contain complex tandem repeats of sequences that can be thousands of base pairs in length.[3] These repeats can be long enough that second generation sequencing reads are not long enough to bridge the repeat, and, as such, determining the location of each repeat in the genome can be difficult.[4] Resolving these tandem repeats can be accomplished by utilizing long third generation sequencing reads, such as those obtained using the PacBio RS DNA sequencer. These sequences are, on average, 10,000-15,000 base pairs in length and are long enough to span most repeated regions.[5] Using a hybrid approach to this process can increase the fidelity of assembling tandem repeats by being able to accurately place them along a linear scaffold and make the process more computationally efficient.
Genome Assembly
Classical Genome Assembly
The term genome assembly refers to the process of taking a large number of DNA fragments that are generated during shotgun sequencing and assembling them into the correct order such as to reconstruct the original genome.[6] Sequencing involves using automated machines to determine the order of nucleic acids in the DNA of interest (the nucleic acids in DNA are adenine, cytosine, guanine and thymine) to conduct genomic analyses involving an organism of interest. The advent of next generation sequencing has presented significant improvements in the speed, accuracy and cost of DNA sequencing and has made the sequencing of entire genomes a feasible process.[7][8] There are many different sequencing technologies that have been developed by various biotechnology companies, each of which produce different sequencing reads in terms of accuracy and read length. Some of these technologies include Roche 454, Illumina, SOLiD, and IonTorrent.[9] These sequencing technologies produce relatively short reads (50-700 bases) and have a high accuracy (>98%). Third generation sequencing include technologies as the PacBio RS system which can produce long reads (maximum of 23kb) but have a relatively low accuracy.[10]
Genome assembly is normally done by one of two methods: assembly using a reference genome as a scaffold,[11] or de novo[12] assembly. The scaffolding approach can be useful if the genome of a similar organism has been previously sequenced. This process involves assembling the genome of interest by comparing it to a known genome or scaffold. De novo genome assembly is used when the genome to be assembled is not similar to any other organisms whose genomes have been previously sequenced. This process is carried out by assembling single reads into contiguous sequences (contigs) which are then extended in the 3’ and 5’ directions by overlapping other sequences. The latter is preferred because it allows for the conservation of more sequences.[13]
The de novo assembly of DNA sequences is a very computationally challenging process and can fall into the NP-hard class of problems if the Hamiltonian-cycle approach is used. This is because millions of sequences must be assembled to reconstruct a genome. Within genomes, there are often tandem repeats of DNA segments that can be thousands of base pairs in length, which can cause problems during assembly.[1]
Although next generation sequencing technology is now capable of producing millions of reads, the assembly of these reads can cause a bottleneck in the entire genome assembly process. As such, extensive research is being done to develop new techniques and algorithms to streamline the genome assembly process and make it a more computationally efficient process and to increase the accuracy of the process as a whole.[10]
Hybrid Genome Assembly

One hybrid approach to genome assembly involves supplementing short, accurate second-generation sequencing data (i.e. from IonTorrent, Illumina or Roche 454) with long less accurate third generation sequencing data (i.e. from PacBio RS) to resolve complex repeated DNA segments.[15] The main limitation of single-molecule third-generation sequencing that prevents it from being used alone is its relatively low accuracy, which causes inherent errors in the sequenced DNA. Using solely second-generation sequencing technologies for genome assembly can miss or lead to the incomplete assembly of important aspects of the genome. Supplementation of third generation reads with short, high-accuracy second generation sequences can overcome these inherent errors and completed crucial details of the genome. This approach has been used to sequence the genomes of some bacterial species including a strain of Vibrio cholerae.[16] Algorithms specific for this type of hybrid genome assembly have been developed, such as the PacBio corrected Reads algorithm.[10]
There are inherent challenges when utilizing sequence reads from various technologies to assemble a sequenced genome; data coming from different sequencers can have different characteristics. An example of this can be seen when using the overlap-layout-consensus (OLC) method of genome assembly, which can be difficult when using reads of substantially different lengths. Currently, this challenge is being overcome by using multiple genome assembly programs.[1] An example of this can be seen in Goldberg et al. where the authors paired 454 reads with Sanger reads. The 454 reads were first assemble using the Newbler assembler (which is optimized to use short reads) generating pseudo reads that were then paired with the longer Sanger reads and assembled using the Celera assembler.[17]
Hybrid genome assembly can also be accomplished using the Eulerian path approach. In this approach, the length of the assembled sequences does not matter as once a k-mer spectrum has been constructed, the lengths of the reads are irrelevant.[1][18]
Practical approaches
Hybrid error correction and de novo assembly of single-molecule sequencing reads
The authors of this study developed a correction algorithm called the PacBio corrected Reads (PBcR) algorithm which is implemented as part of the Celera assembly program.[10] This algorithm calculates an accurate hybrid consensus sequence by mapping higher accuracy short reads (from second generation sequencing technologies) to individual lower accuracy long reads (from third-generation sequencing technologies). This mapping allows for trimming and correction of the long reads to improve the read accuracy from as low as 80% to over 99.9%. In the best example of this application from this paper, the contig size was quintupled when compared to the assemblies using only second-generation reads.[10]
This study offers an improvement over the typical programs and algorithms used to assemble uncorrected PacBio reads. ALLPATHS-LG (another program that can assemble PacBio reads) uses the uncorrected PacBio reads to assist in scaffolding and for the closing of gaps in short sequence assemblies. Due to computational limitations, this approach limits assembly to relatively small genomes (maximum of 10Mbp). The PBcR algorithm allows for the assembly of much larger genomes with higher fidelity and using uncorrected PacBio reads.[10]
This study also shows that using a lower coverage of corrected long reads is similar to using a higher coverage of shorter reads; 13x PBcR data (corrected using 50x Illumina data) was comparable to an assembly constructed using 100x paired-end Illumina reads. The N50 for the corrected PBcR data was also longer than the Illumina data (4.65MBp compared to 3.32 Mbp for the Illumina reads). A similar trend was seen in the sequencing of the Escherichia coli JM221 genome: a 25x PBcR assembly had a N50 triple that of 50x 454 assembly.[10]
Automated finishing of bacterial genomes
This study employed two different methods for hybrid genome assembly: a scaffolding approach that supplemented currently available sequenced contigs with PacBio reads, as well as an error correction approach to improve the assembly of bacterial genomes.[16] The first approach in this study started with high-quality contigs constructed from sequencing reads from second-generation (Illumina and 454) technology. These contigs were supplemented by aligning them to PacBio long reads to achieve linear scaffolds that were gap-filled using PacBio long reads. These scaffolds were then supplemented again, but using PacBio strobe reads (multiple subreads from a single contiguous fragment of DNA [19]) to achieve a final, high-quality assembly. This approach was used to sequence the genome of a strain of Vibrio cholerae that was responsible for a cholera outbreak in Haiti.[16][20]
This study also used a hybrid approach to error-correction of PacBio sequencing data. This was done by utilizing high-coverage Illumina short reads to correct errors in the low-coverage PacBio reads. BLASR (a long read aligner from PacBio) was used in this process. In areas where the Illumina reads could be mapped, a consensus sequence was constructed using overlapping reads in that region.[16]
One area of the genome where the use of the long PacBio reads was especially helpful was the ribosomal operon. This region is usually greater than 5kb in size and occurs seven time throughout the genome with an average identity ranging from 98.04% to 99.94%. Resolving these regions using only short second generation reads would be very difficult but the use of long third generation reads makes the process much more efficient. Utilization of the PacBio reads allowed for unambiguous placement of the complex repeated along the scaffold.[16]
Using only short reads
This study employs a hybrid genome assembly approach that only uses sequencing reads generated using SOLiD sequencing (a second-generation sequencing technology).[13] The genome of C. pseudotuberculosis was assembled twice: once using a classical reference genome approach, and once using a hybrid approach. The hybrid approach consisted of three contiguous steps. Firstly, contigs were generated de novo, secondly, the contigs were ordered and concatenated into supercontigs, and, thirdly, the gaps between contigs were closed using an iterative approach. The initial de novo assembly of contigs was achieved in parallel using Velvet, which assembles contigs by manipulating De Bruijn graphs, and Edena, which is an OLC-based assembler[13]
Comparing the assembly constructed using the hybrid approach to the assembly created using the traditional reference genome approach showed that, with the availability of a reference genome, it is more beneficial to utilize an hybrid de novo assembly strategy as it preserves more genome sequences.[13]
Using high throughput short and long reads
The authors of this paper present Cerulean, a hybrid genome assembly program that differs from traditional hybrid assembly approaches.[21] Normally, hybrid assembly involved mapping short high quality reads to long low quality reads, but this still introduces errors in the assembled genomes. This process is also computationally expensive and require a large amount of running time, even for relatively small bacterial genomes.[21]
Cerulean, unlike other hybrid assembly approaches, doesn’t use the short reads directly, instead it uses an assembly graph that is created in a similar manner to the OLC method or the De Bruijn method. This graph is used to assemble a skeleton graph, which only uses long contigs with the edges of the graph representing the putative genomic connection between the contigs. The skeleton graph is a simplified version of a typical De Bruijn graph, which means that unambiguous assembly using the skeleton graph is more favourable than traditional methods.[21]
This method was tested by assembling the genome of an ‘’Escherichia coli’’ strain. First, short reads were assembled using the ABySS assembler. These reads were then mapped to the long reads using BLASR. The results from the ABySS assembly were used to create the assembly graph, which were used to generate scaffolds using the filtered BLASR data . The advantages of cerulean are that it requires minimal resources and results in assembled scaffolds with high accuracy. These characteristics make it better suited for up-scaling to be used on larger eukaryotic genomes, but the efficiency of cerulean when applied to larger genomes remains to be verified.[21]
Future prospectives
The current challenges in genome assembly are related to the limitation of modern sequencing technologies. Advances in sequencing technology aim to develop systems that are able to produce long sequencing reads with very high fidelity but, at this point, these two things are mutually exclusive.[1] The advent of third-generation sequencing technology is expanding the limits of genomic research as the cost of generating high quality sequencing data is decreasing.[22]
The idea of using multiple sequencing technologies to facilitate genome assembly may become an idea of the past as the quality of long sequencing reads (hundreds or thousands of base pairs) approaches and exceeds the quality of current second generation sequencing reads. The computational difficulties that are encountered during genome assembly will also become a concept of the past as computation efficiency and performance increases. The development of more efficient sequencing algorithms and assembly programs is needed to develop more effective assembly approaches that can tandemly incorporate sequencing reads from multiple technologies.
Many of the current limitations in genomic research revolve around the ability to produce large amounts of high quality sequencing data and to assemble entire genomes of organisms of interest. Developing more effective hybrid genome assembly strategies is taking the next step in advancing sequence assembly technology and these strategies are guaranteed to become more effective as more powerful technologies emerge.
References
- Pop, M. (2009). Genome assembly reborn: recent computational challenges. Brief Bioinform, 10(4), 354-366. doi: 10.1093/bib/bbp026.
- Pellicer, Jaume, Fay, Michael F., & Leitch, Ilia J. (2010). The largest eukaryotic genome of them all? Botanical Journal of the Linnean Society, 164(1), 10-15. doi: 10.1111/j.1095-8339.2010.01072.x
- Alkan, C., Sajjadian, S., & Eichler, E. (2011). Limitations of next-generation genome sequence assembly. Nature Methods, 8.
- Koren, S., Harhay, G., Smith, P., Bono, J., Harhay, D., Mcvey, S., . . . Phillippy, A. (2013). Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biology.
- http://blog.pacificbiosciences.com/2014/10/new-chemistry-boosts-average-read.html
- Motahari, A. S., Bresler, G., & Tse, D. N. C. (2013). Information Theory of DNA Shotgun Sequencing. IEEE Transactions on Information Theory, 59(10), 6273-6289. doi: 10.1109/tit.2013.2270273
- Mardis, E. R. (2008). Next-generation DNA sequencing methods. Annu Rev Genom Hum Genet, 9, 387-402. doi: 10.1146/annurev.genom.9.081307.164359
- DiGuistini, S., Liao, N., Platt, D., Robertson, G., Siedel, M., Chan, S., . . . Jones, S. J. M. (2009). De novo sequence assembly of a filamentous fungus using Sanger, 454 and Illumina sequence data. Genome Biology, 10.
- Glenn, T. (2011). Field guide to next-generation DNA sequencers. Molecular Ecology Resources, 11.
- Koren, S., Schatz, M. C., Walenz, B. P., Martin, J., Howard, J. T., Ganapathy, G., . . . Phillippy, A. M. (2012). Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature Biotechnology, 30(7), 692-+. doi: 10.1038/nbt.2280
- Kim, P. G., Cho, H. G., & Park, K. (2008). A scaffold analysis tool using mate-pair information in genome sequencing. Journal of Biomedicine and Biotechnology. doi: 10.1155/2008/675741
- Ham, J. S., Kwak, W., Chang, O. K., Han, G. S., Jeong, S. G., Seol, K. H., . . . Kim, H. (2013). De Novo Assembly and Comparative Analysis of the Enterococcus faecalis Genome (KACC 91532) from a Korean Neonate. Journal of Microbiology and Biotechnology, 23(7), 966-973. doi: 10.4014/jmb.1303.03045
- Cerdeira, L. T., Carneiro, A. R., Ramos, R. T. J., de Almeida, S. S., D'Afonseca, V., Schneider, M. P. C., . . . Silva, A. (2011). Rapid hybrid de novo assembly of a microbial genome using only short reads: Corynebacterium pseudo tuberculosis I19 as a case study. Journal of Microbiological Methods, 86(2), 218-223. doi: 10.1016/j.mimet.2011.05.008.
- Wang, Y., Yu, Y., Pan, B., Hao, P., Li, Y., Shao, Z., . . . Li, X. (2012). Optimizing hybrid assembly of next-generation sequence data from Enterococcus faecium: a microbe with highly divergent genome. BMC Syst Biol, 6 Suppl 3, S21. doi: 10.1186/1752-0509-6-S3-S21
- English, A. C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J. X., . . . Gibbs, R. A. (2012). Mind the Gap: Upgrading Genomes with Pacific Biosciences RS Long-Read Sequencing Technology. PLoS ONE, 7(11). doi: 10.1371/journal.pone.0047768
- Bashir, A., Klammer, A. A., Robins, W. P., Chin, C. S., Webster, D., Paxinos, E., . . . Schadt, E. E. (2012). A hybrid approach for the automated finishing of bacterial genomes. Nature Biotechnology, 30(7), 701-+. doi: 10.1038/nbt.2288
- Goldberg, S. M., Johnson, J., Busam, D., Feldblyum, T., Ferriera, S., Friedman, R., . . . Venter, J. C. (2006). A Sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc Natl Acad Sci U S A, 103(30), 11240-11245. doi: 10.1073/pnas.0604351103
- Pevzner, P. A., Tang, H., & Waterman, M. S. (2001). An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci U S A, 98(17), 9748-9753. doi: 10.1073/pnas.171285098
- Ritz, Anna, Bashir, Ali, & Raphael, Benjamin J. (2010). Structural variation analysis with strobe reads. Bioinformatics, 26(10), 1291-1298. doi: 10.1093/bioinformatics/btq153
- Abrams, J. Y., Copeland, J. R., Tauxe, R. V., Date, K. A., Belay, E. D., Mody, R. K., & Mintz, E. D. (2013). Real-time modelling used for outbreak management during a cholera epidemic, Haiti, 2010-2011. Epidemiology and Infection, 141(6), 1276-1285.
- Deshpande, V., Fung, E., Pham, S., & Bafna, V. (2013). Cerulean: A hybrid assembly using high throughput short and long reads. Algorithms in Bioinformatics, 8126, 349-363.
- http://www.ddw-online.com/enabling-technologies/p211492-dna-sequencing:towards-the-third-generation-and-beyondspring-13.html
External links
Hybrid Error Correction and De Novo Assembly of Single-Molecule Sequencing Reads
Virtual Poster: Hybrid Genome Assembly of a Nocturnal Lemur
National Center for Biotechnology Information: Genome Assembly