Genomic phylostratigraphy
Genomic phylostratigraphy is a novel genetic statistical method developed in order to date the origin of specific genes by looking at its homologs across species. It was first developed by Ruđer Bošković Institute in Zagreb, Croatia.[1] The system links genes to their founder gene, allowing us to then determine their age. This could in turn help us better understand many evolutionary processes.
Method
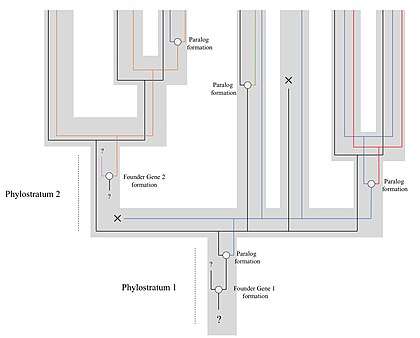
This technique relies on the assumption that the diversity of the genome is not only due to gene duplications but also to continuous frequent de novo gene births. These genes (called "founder genes") would form from non-genic DNA sequences, as well as from changes in reading frame (or other ways of arising from within existing genes), or even from very rapid evolution of the protein that would modify the sequence beyond recognition.[2] These new genes would at first have high evolutionary rates that would then slow down with time, allowing us to recognise their lineage in their descendants.[1] The founder genes can then be put in a specific phylostratum. The phylostratum is represented as the clade that includes all the genes that derive from the same founder gene, signifying that this gene was formed in the common ancestor of this clade (e.g. Arthropoda, Mammalia, Metazoa, etc.). Positioning these founder genes and their descendants on different phylostrata can allow us to age them. This can then be used to analyse the origin of certain functions of proteins and developmental processes on a macroevolutionary scale, by observing connections between certain genes as well.
The original method for genomic phylostratigraphy involves the use of a BLAST sequence similarity search with a 10−3 E-value cut off. The genes deemed similar enough in sequence are gathered and the clade englobing all the taxa represented by those genes is determined. This clade then becomes the phylostratum of these genes. By determining the common ancestor of this clade, we can hence give an age to the founder gene and all its descendants. Applying the process on a genome-wide scale can then allow us to detect patterns of founder genes births and infer the role of certain genes involved in clade-specific developmental processes and physiological pathways, and the origin of those traits. The developers of the method gave in the original paper[1] an example how to exploit this system in practice using Drosophila. They gathered 13,000 genes for which they determined the founder genes, regrouping them in their respective phylostrata. They also segregated the families of genes depending on whether they were mainly expressed in either of the three germ layers (endoderm, mesoderm, ectoderm). By studying the frequencies of expression of genes in those different phylostrata, they were able to hypothetically pinpoint the possible original formation of those germ layers to specific periods and ancestral organisms in evolutionary history. Since its invention, genomic phylostratigraphy has been regularly used by this research team[3] as well as others,[4] notably in an attempt to determine the origin of cancer genes, seemingly showing a strong link between a peak in the formation of cancer genes and the transition to multicellular organisms, a connection which had been previously hypothesised and is hence further supported by phylostratigraphy. As its use has grown, the method has been assessed and enhanced on multiple occasions, and programs that run it automatically and more efficiently have been developed.[2]
Criticism
Albeit it being now used frequently by the scientific community, genomic phylostratigraphy has also received some criticism for being too inaccurate for its measurements to be trustworthy. First of all, according to some authors precision lacks in the assumptions.[5][6] It is erroneous to assume for example that all species beyond the organism of focus share the same protein evolutionary rate, which isn't true as it varies depending on cell cycle speeds, leading to problems in setting the limits of BLAST error to englobe all proteins originated from the same founder gene. Another point is that the BLAST search assumes that protein evolutionary rates is constant at all its sites, which is also false. Lastly, it could be said that the model does not account correctly for gene duplications, as well as gene losses: the changes in evolutionary rates caused by gene duplications due to new functional changes would increase BLAST error rates, and gene loss in taxa distant to the one studied could lead to great underestimations in the calculated gene age and phylostratum of founder genes compared to their true values. However, rather than demanding to simply abandon the method, critics have been trying to work at refining it from its original state, by introducing other potential mathematical formulas or sequence searching tools,[7] although the Ruđer Bošković Institute has replied to such criticism claiming their original approach was valid and did not need to be extensively revised.[8] This debate is also included as part of the wider discussion on the importance of de novo gene births in creating genetic diversity, in which genomic phylostratigraphy supports that they do hold a strong effect, in a way that it can only be widely accepted or refuted once the latter dilemma has been resolved.
References
- Domazet-Lošo, Tomislav; Brajković, Josip; Tautz, Diethard (November 2007). "A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages". Trends in Genetics. 23 (11): 533–539. doi:10.1016/j.tig.2007.08.014. PMID 18029048.
- Arendsee, Zebulun; Li, Jing; Singh, Urminder; Seetharam, Arun; Dorman, Karin; Wurtele, Eve Syrkin (2018-07-03). "phylostratr: A framework for phylostratigraphy". bioRxiv. doi:10.1101/360164.
- Domazet-Lošo, Tomislav; Tautz, Diethard (December 2010). "Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa". BMC Biology. 8 (1): 66. doi:10.1186/1741-7007-8-66. ISSN 1741-7007. PMC 2880965. PMID 20492640.
- Zhang, Luoyan; Tan, Yi; Fan, Shoujin; Zhang, Xuejie; Zhang, Zhen (December 2019). "Phylostratigraphic analysis of gene co-expression network reveals the evolution of functional modules for ovarian cancer". Scientific Reports. 9 (1): 2623. doi:10.1038/s41598-019-40023-9. ISSN 2045-2322. PMC 6384884. PMID 30796309.
- Moyers, Bryan A.; Zhang, Jianzhi (2015-01-01). "Phylostratigraphic Bias Creates Spurious Patterns of Genome Evolution". Molecular Biology and Evolution. 32 (1): 258–267. doi:10.1093/molbev/msu286. ISSN 0737-4038. PMC 4271527. PMID 25312911.
- Casola, Claudio (2018-10-22). "From de novo to 'de nono': The majority of novel protein coding genes identified with phylostratigraphy are old genes or recent duplicates". Genome Biology and Evolution. 10 (11): 2906–2918. doi:10.1093/gbe/evy231. ISSN 1759-6653. PMC 6239577. PMID 30346517.
- Moyers, Bryan A; Zhang, Jianzhi (2018-08-01). Martin, Bill (ed.). "Toward Reducing Phylostratigraphic Errors and Biases". Genome Biology and Evolution. 10 (8): 2037–2048. doi:10.1093/gbe/evy161. ISSN 1759-6653. PMC 6105108. PMID 30060201.
- Domazet-Lošo, Tomislav; Carvunis, Anne-Ruxandra; Mar Albà, M.; Sebastijan Šestak, Martin; Bakarić, Robert; Neme, Rafik; Tautz, Diethard (2017-01-12). "No evidence for phylostratigraphic bias impacting inferences on patterns of gene emergence and evolution". Molecular Biology and Evolution. 34 (4): 843–856. doi:10.1093/molbev/msw284. ISSN 0737-4038. PMC 5400388. PMID 28087778.