Genome-wide CRISPR-Cas9 knockout screens
Genome-wide CRISPR-Cas9 knockout screens aim to elucidate the relationship between genotype and phenotype by ablating gene expression on a genome-wide scale and studying the resulting phenotypic alterations. The approach utilises the CRISPR-Cas9 gene editing system, coupled with libraries of single guide RNAs (sgRNAs), which are designed to target every gene in the genome. Over recent years, the genome-wide CRISPR screen has emerged as a powerful tool for performing large-scale loss-of-function screens, with low noise, high knockout efficiency and minimal off-target effects.
History
Early studies in Caenorhabditis elegans[1] and Drosophila melanogaster[2][3] saw large-scale, systematic loss of function (LOF) screens performed through saturation mutagenesis, demonstrating the potential of this approach to characterise genetic pathways and identify genes with unique and essential functions. The saturation mutagenesis technique was later applied in other organisms, for example zebrafish[4][5] and mice.[6][7]
Targeted approaches for gene knockdown emerged in the 1980s with techniques such as homologous recombination,[8][9] trans-cleaving ribozymes,[10][11] and antisense technologies.[12][13]
By the year 2000, RNA interference (RNAi) technology had emerged as a fast, simple, and inexpensive technique for targeted gene knockdown, and was routinely being used to study in vivo gene function in C. elegans.[14][15][16][17] Indeed, in the span of only a few years following its discovery by Fire et al. (1998),[18] almost all of the ~19,000 genes in C. elegans had been analysed using RNAi-based knockdown.[19]
The production of RNAi libraries facilitated the application of this technology on a genome-wide scale, and RNAi-based methods became the predominant approach for genome-wide knockdown screens.
Nevertheless, RNAi-based approaches to genome-wide knockdown screens have their limitations. For one, the high off-target effects cause issues with false-positive observations.[20][21] Additionally, because RNAi reduces gene expression at the post-transcriptional level by targeting RNA, RNAi-based screens only result in partial and short-term suppression of genes. Whilst partial knockdown may be desirable in certain situations, a technology with improved targeting efficiency and fewer off-target effects was needed.
Since initial identification as a prokaryotic adaptive immune system,[22] the bacterial type II clustered regularly interspaced short palindrome repeats (CRISPR)/Cas9 system has become a simple and efficient tool for generating targeted LOF mutations.[23] It has been successfully applied to edit human genomes, and has started to displace RNAi as the dominant tool in mammalian studies.[24] In the context of genome-wide knockout screens, recent studies have demonstrated that CRISPR/Cas9 screens are able to achieve highly efficient and complete protein depletion, and overcome the off-target issues seen with RNAi screens.[25][26] In summary, the recent emergence of CRISPR-Cas9 has dramatically increased our ability to perform large-scale LOF screens. The versatility and programmability of Cas9, coupled with the low noise, high knockout efficiency and minimal off-target effects, have made CRISPR the platform of choice for many researchers engaging in gene targeting and editing.[24][27]
Methods
CRISPR/Cas9 Loss of function
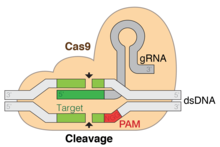
The clustered regularly interspaced short palindrome repeats (CRISPR)/Cas9 system is a gene-editing technology that can introduce double-strand breaks (DSBs) at a target genomic locus. By using a single guide RNA (sgRNA), the endonuclease Cas9 can be delivered to a specific DNA sequence where it cleaves the nucleotide chain.[28] The specificity of the sgRNA is determined by a 20-nt sequence, homologous to the genomic locus of interest, and the binding to Cas9 is mediated by a constant scaffold region of the sgRNA. The desired target site must be immediately followed (5’ to 3’) by a conserved 3 nucleotide protospacer adjacent motif (PAM).[29][30] In order to repair the DSBs, the cell may use the highly error prone non-homologous end joining, or homologous recombination. By designing suitable sgRNAs, planned insertions or deletions can be introduced into the genome. In the context of genome-wide LOF screens, the aim is to cause gene disruption and knockout.
sgRNA libraries
Constructing a Library
To perform CRISPR knockouts on a genome-wide scale, collections of sgRNAs known as sgRNA libraries, or CRISPR knockout libraries, must be generated. The first step in creating a sgRNA library is to identify genomic regions of interest based on known sgRNA targeting rules.[31] For example, sgRNAs are most efficient when targeting the coding regions of genes and not the 5’ and 3’ UTRs. Conserved exons present as attractive targets, and position relative to the transcription start site should be considered.[31] Secondly, all the possible PAM sites are identified and selected for.[31] On- and off-target activity should be analysed, as should GC content, and homopolymer stretches should be avoided.[31] The most commonly used Cas9 endonuclease, derived from Streptococcus pyogenes, recognises a PAM sequence of NGG.[32]
Furthermore, specific nucleotides appear to be favoured at specific locations. Guanine is strongly favoured over cytosine on position 20 right next to the PAM motif, and on position 16 cytosine is preferred over guanine.[33] For the variable nucleotide in the NGG PAM motif, it has been shown that cytosine is preferred and thymine disfavoured.[33] With such criteria taken into account, the sgRNA library is computationally designed around the selected PAM sites.[31][33][34]
Multiple sgRNAs (at least 4-6) should be created against every single gene to limit false-positive detection, and negative control sgRNAs with no known targets should be included.[31][33] The sgRNAs are then created by in situ synthesis, amplified by PCR, and cloned into a vector delivery system.
Existing libraries
Developing a new sgRNA library is a laborious and time-consuming process. In practice, researchers may select an existing library depending on their experimental purpose and cell lines of interest. As of February 2020, the most widely used resources for genome-wide CRISPR knockout screens have been the two Genome-Scale CRISPR Knock-Out (GeCKO) libraries created by the Zhang lab.[35] Available through addgene, these lentiviral libraries respectively target human and mouse exons, and both are available as a one-vector system (where the sgRNAs and Cas9 are present on the same plasmid) or as a two-vector system (where the sgRNAs and Cas9 are present on separate plasmids). Each library is delivered as two half-libraries, allowing researchers to screen with 3 or 6 sgRNAs/gene.[36]
Aside from GeCKO, a number of other CRISPR libraries have been generated and made available through addgene. The Sabatini & Lander labs currently have 7 separate human and mouse libraries, including targeted sublibraries for distinct subpools such as kinases and ribosomal genes (Addgene #51043–51048). Further, improvements to the specificity of sgRNAs have resulted in ‘second generation’ libraries, such as the Brie (Addgene #73632) and Brunello (Addgene #73178) libraries generated by the Doench and Root labs, and the Toronto knockout (TKO) library (Addgene #1000000069) generated by the Moffat lab.[36]
Lentiviral vectors
Targeted gene knockout using CRISPR/Cas9 requires the use of a delivery system to introduce the sgRNA and Cas9 into the cell. Although a number of different delivery systems are potentially available for CRISPR,[37][38] genome-wide loss-of-function screens are predominantly carried out using third generation lentiviral vectors.[35][39][40] These lentiviral vectors are able to efficiently transduce a broad range of cell types and stably integrate into the genome of dividing and non-dividing cells.[41][42] Third generation lentiviral particles are produced by co-transfecting 293T human embryonic kidney (HEK) cells with:
- two packaging plasmids, one encoding Rev and the other Gag and Pol;
- an interchangeable envelope plasmid that encodes for an envelope glycoprotein of another virus (most commonly the G protein of vesicular stomatitis virus (VSV-G));
- one or two (depending on the applied library) transfer plasmids, encoding for Cas9 and sgRNA, as well as selection markers.[35][43][44]
The lentiviral particle-containing supernatant is harvested, concentrated and subsequently used to infect the target cells.[45] The exact protocol for lentiviral production will vary depending on the research aim and applied library.[35][43][44] If a two vector-system is used, for example, cells are sequentially transduced with Cas9 and sgRNA in a two-step procedure.[35][44] Although more complex, this has the advantage of a higher titre for the sgRNA library virus.[35]
Phenotypic selection
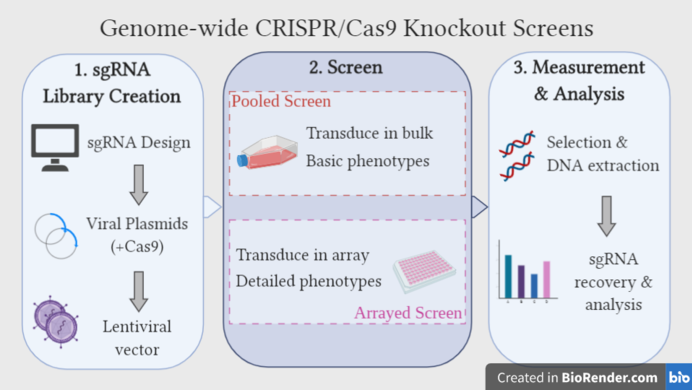
In general, there are two different formats of genome-wide CRISPR knockout screens: arrayed and pooled. In an arrayed screen, each well contains a specific and known sgRNA targeting a specific gene.[46] Since the sgRNA responsible for each phenotype is known based on well location, phenotypes can be identified and analysed without requiring genetic sequencing. This format allows for the measurement of more specific cellular phenotypes, perhaps by fluorescence or luminescence, and allows researchers to use more library types and delivery methods.[46] For large-scale LOF screens, however, arrayed formats are considered low-efficiency, and expensive in terms of financial and material resources because cell populations have to be isolated and cultured individually.[46]
In a pooled screen, cells grown in a single vessel are transduced in bulk with viral vectors collectively containing the entire sgRNA library. To ensure that the amount of cells infected by more than one sgRNA-containing particle is limited, a low multiplicity of infection (MOI) (typically 0.3-0.6) is used.[46][47] Evidence so far has suggested that each sgRNA should be represented in a minimum of 200 cells.[48][23] Transduced cells will be selected for, followed by positive or negative selection for the phenotype of interest, and genetic sequencing will be necessary to identify the integrated sgRNAs.[46]
Next-generation sequencing & hit analysis
Following phenotypic selection, genomic DNA is extracted from the selected clones, alongside a control cell population.[23][46][49] In the most common protocols for genome-wide knockouts, a 'Next-generation sequencing (NGS) library' is created by a two step polymerase chain reaction (PCR).[23][46] The first step amplifies the sgRNA region, using primers specific to the lentiviral integration sequence, and the second step adds Illumina i5 and i7 sequences.[23] NGS of the PCR products allows the recovered sgRNAs to be identified, and a quantification step can be used to determine the relative abundance of each sgRNA.[23]
The final step in the screen is to computationally evaluate the significantly enriched or depleted sgRNAs, trace them back to their corresponding genes, and in turn determine which genes and pathways could be responsible for the observed phenotype. Several algorithms are currently available for this purpose, with the most popular being the Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) method.[50] Developed specifically for CRISPR/Cas9 knockout screens in 2014, MAGeCK demonstrated better performance compared with alternative algorithms at the time,[50] and has since demonstrated robust results and high sensitivity across different experimental conditions.[51] As of 2015, the MAGeCK algorithm has been extended to introduce quality control measurements, and account for the previously overlooked sgRNA knockout efficiency.[51] A web-based visualisation tool (VISPR) was also integrated, allowing users to interactively explore the results, analysis, and quality controls.[51]
Applications
Cellular signalling mechanisms
Over recent years, the genome-wide CRISPR screen has emerged as a powerful tool for studying the intricate networks of cellular signalling.[52] Cellular signalling is essential for a number of fundamental biological processes, including cell growth, proliferation, differentiation, and apoptosis.
One practical example is the identification of genes required for proliferative signalling in cancer cells. Cells are transduced with a CRISPR sgRNA library, and studied for growth over time. By comparing sgRNA abundance in selected cells to a control, one can identify which sgRNAs become depleted and in turn which genes may be responsible for the proliferation defect. Such screens have been used to identify cancer-essential genes in acute myeloid leukemia[53] and neuroblastoma,[54] and to describe tumour-specific differences between cancer cell lines.[55]
Identifying synthetic lethal partners
Targeted cancer therapies are designed to target the specific genes, proteins, or environments contributing to tumour cell growth or survival. After a period of prolonged treatment with these therapies, however, tumour cells may develop resistance. Although the mechanisms behind cancer drug resistance are poorly understood, potential causes include: target alteration, drug degradation, apoptosis escape, and epigenetic alterations.[56] Resistance is well-recognised and poses a serious problem in cancer management.
To overcome this problem, a synthetic lethal partner can be identified. Genome-wide LOF screens using CRISPR-Cas9 can be used to screen for synthetic lethal partners.[57] For this, a wild-type cell line and a tumour cell line containing the resistance-causing mutation are transduced with a CRISPR sgRNA library. The two cell lines are cultivated, and any under-represented or dead cells are analysed to identify potential synthetic lethal partner genes. A recent study by Hinze et al. (2019)[58] used this method to identify a synthetic lethal interaction between the chemotherapy drug asparaginase and two genes in the Wnt signalling pathway NKD2 and LGR6.
Host dependency factors for viral infection
Due to their small genomes and limited number of encoded proteins, viruses exploit host proteins for entry, replication, and transmission. Identification of such host proteins, also termed host dependency factors (HDFs), is particularly important for identifying therapeutic targets. Over recent years, many groups have successfully used genome-wide CRISPR/Cas9 as a screening strategy for HDFs in viral infections.[59]
One example is provided by Marceau et al. (2017),[60] who aimed to dissect the host factors associated with dengue and hepatitis C (HCV) infection (two viruses in family Flaviviridae). ELAVL1, an RNA-binding protein encoded by the ELAVL1 gene, was found to be a critical receptor for HCV entry, and a remarkable divergence in host dependency factors was demonstrated between the two flaviviridae.[60]
Further applications
Additional reported applications of genome-wide CRISPR screens include the study of: mitochondrial metabolism,[61] bacterial toxin resistance,[62] genetic drivers of metastasis,[63] cancer drug resistance,[64] West Nile virus-induced cell death,[65] and immune cell gene networks.[66][67]
Limitations
This section will specifically address genome-wide CRISPR screens. For a review of CRISPR limitations see Lino et al. (2018)[38]
The sgRNA library
Genome-wide CRISPR screens will ultimately be limited by the properties of the chosen sgRNA library. Each library will contain a different set of sgRNAs, and average coverage per gene may vary. Currently available libraries tend to be biased towards sgRNAs targeting early (5’) protein-coding exons, rather than those targeting the more functional protein domains.[58] This problem was highlighted by Hinze et al. (2019),[58] who noted that genes associated with asparaginase sensitivity failed to score in their genome-wide screen of asparaginase-resistant leukemia cells.
If an appropriate library is not available, creating and amplifying a new sgRNA library is a lengthy process which may take many months. Potential challenges include: (i) effective sgRNA design; (ii) ensuring comprehensive sgRNA coverage throughout the genome; (iii) lentiviral vector backbone design; (iv) producing sufficient amounts of high-quality lentivirus; (v) overcoming low transformation efficiency; (vi) proper scaling of the bacterial culture.[68]
Maintaining cellular sgRNA coverage
One of the largest hurdles for genome-wide CRISPR screening is ensuring adequate coverage of the sgRNA library across the cell population.[23] Evidence so far has suggested that each sgRNA should be represented and maintained in a minimum of 200-300 cells.[23][48]
Considering that the standard protocol uses a multiplicity of infection of ~0.3, and a transduction efficiency of 30-40%[44][23] the number of cells required to produce and maintain suitable coverage becomes very large. By way of example, the most popular human sgRNA library is the GeCKO v2 library created by the Zhang lab;[30] it contains 123,411 sgRNAs. Studies using this library commonly transduce more than 1x108 cells [58][59][69]
As CRISPR continues to exhibit low noise and minimal off-target effects, an alternative strategy is to reduce the number of sgRNAs per gene for a primary screen. Less stringent cut-offs are used for hit selection, and additional sgRNAs are later used in a more specific secondary screen. This approach is demonstrated by Doench et al. (2016),[33] who found that >92% of genes recovered using the standard protocol were also recovered using fewer sgRNAs per gene. They suggest that this strategy could be useful in studies where scale-up is prohibitively costly.
Lentiviral limitations
Lentiviral vectors have certain general limitations. For one, it is impossible to control where the viral genome integrates into the host genome, and this may affect important functions of the cell. Vannucci et al.[70] provide an excellent review of viral vectors along with their general advantages and disadvantages. In the specific context of genome-wide CRISPR screens, producing and transducing the lentiviral particles is relatively laborious and time consuming, taking about two weeks in total.[44] Additionally, because the DNA integrates into the host genome, lentiviral delivery leads to long-term expression of Cas9, potentially leading to off-target effects.
Arrayed vs pooled screens
In an arrayed screen, each well contains a specific and known sgRNA targeting a specific gene. Arrayed screens therefore allow for detailed profiling of a single cell, but are limited by high costs and the labour required to isolate and culture the high number of individual cell populations.[46] Conventional pooled CRISPR screens are relatively simple and cost effective to perform, but are limited to the study of the entire cell population. This means that rare phenotypes may be more difficult to identify, and only crude phenotypes can be selected for e.g. cell survival, proliferation, or reporter gene expression.
Future Directions
CRISPR + single cell RNA-seq
Emerging technologies are aiming to combine pooled CRISPR screens with the detailed resolution of massively parallel single-cell RNA-sequencing (RNA-seq). Studies utilising “CRISP-seq”,[71] “CROP-seq”,[72] and “PERTURB-seq”[73] have demonstrated rich genomic readouts, accurately identifying gene expression signatures for individual gene knockouts in a complex pool of cells. These methods have the added benefit of producing transcriptional profiles of the sgRNA-induced cells.
References
- Brenner S (May 1974). "The genetics of Caenorhabditis elegans". Genetics. 77 (1): 71–94. PMC 1213120. PMID 4366476.
- Gans M, Audit C, Masson M (December 1975). "Isolation and characterization of sex-linked female-sterile mutants in Drosophila melanogaster". Genetics. 81 (4): 683–704. PMC 1213428. PMID 814037.
- Nüsslein-Volhard C, Wieschaus E (October 1980). "Mutations affecting segment number and polarity in Drosophila". Nature. 287 (5785): 795–801. Bibcode:1980Natur.287..795N. doi:10.1038/287795a0. PMID 6776413.
- Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, et al. (December 1996). "The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio". Development. 123: 1–36. PMID 9007226.
- Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, et al. (December 1996). "A genetic screen for mutations affecting embryogenesis in zebrafish". Development. 123: 37–46. PMID 9007227.
- Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, et al. (August 2000). "A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse". Nature Genetics. 25 (4): 440–3. doi:10.1038/78140. PMID 10932191.
- Kasarskis A, Manova K, Anderson KV (June 1998). "A phenotype-based screen for embryonic lethal mutations in the mouse". Proceedings of the National Academy of Sciences of the United States of America. 95 (13): 7485–90. Bibcode:1998PNAS...95.7485K. doi:10.1073/pnas.95.13.7485. PMC 22659. PMID 9636176.
- Gossler A, Doetschman T, Korn R, Serfling E, Kemler R (December 1986). "Transgenesis by means of blastocyst-derived embryonic stem cell lines". Proceedings of the National Academy of Sciences of the United States of America. 83 (23): 9065–9. Bibcode:1986PNAS...83.9065G. doi:10.1073/pnas.83.23.9065. PMC 387075. PMID 3024164.
- Capecchi MR (June 1989). "Altering the genome by homologous recombination". Science. 244 (4910): 1288–92. Bibcode:1989Sci...244.1288C. doi:10.1126/science.2660260. PMID 2660260.
- Zaug AJ, Grosshans CA, Cech TR (December 1988). "Sequence-specific endoribonuclease activity of the Tetrahymena ribozyme: enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme-substrate complexes". Biochemistry. 27 (25): 8924–31. doi:10.1021/bi00425a008. PMID 3069131.
- Cameron FH, Jennings PA (December 1989). "Specific gene suppression by engineered ribozymes in monkey cells". Proceedings of the National Academy of Sciences of the United States of America. 86 (23): 9139–43. Bibcode:1989PNAS...86.9139C. doi:10.1073/pnas.86.23.9139. PMC 298449. PMID 2556702.
- Ecker JR, Davis RW (August 1986). "Inhibition of gene expression in plant cells by expression of antisense RNA". Proceedings of the National Academy of Sciences of the United States of America. 83 (15): 5372–6. Bibcode:1986PNAS...83.5372E. doi:10.1073/pnas.83.15.5372. PMC 386288. PMID 16593734.
- Toulmé JJ, Hélène C (December 1988). "Antimessenger oligodeoxyribonucleotides: an alternative to antisense RNA for artificial regulation of gene expression--a review". Gene. 72 (1–2): 51–8. doi:10.1016/0378-1119(88)90127-8. PMID 2468575.
- Chase D, Serafinas C, Ashcroft N, Kosinski M, Longo D, Ferris DK, Golden A (January 2000). "The polo-like kinase PLK-1 is required for nuclear envelope breakdown and the completion of meiosis in Caenorhabditis elegans". Genesis. 26 (1): 26–41. doi:10.1002/(sici)1526-968x(200001)26:1<26::aid-gene6>3.0.co;2-o. PMID 10660671.
- Kawano T, Fujita M, Sakamoto H (July 2000). "Unique and redundant functions of SR proteins, a conserved family of splicing factors, in Caenorhabditis elegans development". Mechanisms of Development. 95 (1–2): 67–76. doi:10.1016/s0925-4773(00)00339-7. PMID 10906451.
- Powers J, Bossinger O, Rose D, Strome S, Saxton W (October 1998). "A nematode kinesin required for cleavage furrow advancement". Current Biology. 8 (20): 1133–6. doi:10.1016/s0960-9822(98)70470-1. PMC 3209536. PMID 9778533.
- Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, et al. (August 2000). "Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes". Cell. 102 (3): 279–91. doi:10.1016/s0092-8674(00)00034-9. PMID 10975519.
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (February 1998). "Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans". Nature. 391 (6669): 806–11. Bibcode:1998Natur.391..806F. doi:10.1038/35888. PMID 9486653.
- Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK (December 2003). "RNA interference: biology, mechanism, and applications". Microbiology and Molecular Biology Reviews. 67 (4): 657–85. doi:10.1128/mmbr.67.4.657-685.2003. PMC 309050. PMID 14665679.
- Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, King RW (February 2012). "A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens". Nature Methods. 9 (4): 363–6. doi:10.1038/nmeth.1898. PMC 3482495. PMID 22343343.
- Echeverri CJ, Beachy PA, Baum B, Boutros M, Buchholz F, Chanda SK, et al. (October 2006). "Minimizing the risk of reporting false positives in large-scale RNAi screens". Nature Methods. 3 (10): 777–9. doi:10.1038/nmeth1006-777. PMID 16990807.
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. (March 2007). "CRISPR provides acquired resistance against viruses in prokaryotes". Science. 315 (5819): 1709–12. Bibcode:2007Sci...315.1709B. doi:10.1126/science.1138140. hdl:20.500.11794/38902. PMID 17379808.
- Yau EH, Rana TM (2018). "Next-Generation Sequencing of Genome-Wide CRISPR Screens". Next Generation Sequencing. Methods in Molecular Biology. 1712. pp. 203–216. doi:10.1007/978-1-4939-7514-3_13. ISBN 978-1-4939-7512-9. PMC 6089254. PMID 29224076.
- Boettcher M, McManus MT (May 2015). "Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR". Molecular Cell. 58 (4): 575–85. doi:10.1016/j.molcel.2015.04.028. PMC 4441801. PMID 26000843.
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. (October 2014). "Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation". Cell. 159 (3): 647–61. doi:10.1016/j.cell.2014.09.029. PMC 4253859. PMID 25307932.
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. (February 2013). "Multiplex genome engineering using CRISPR/Cas systems". Science. 339 (6121): 819–23. Bibcode:2013Sci...339..819C. doi:10.1126/science.1231143. PMC 3795411. PMID 23287718.
- Evers B, Jastrzebski K, Heijmans JP, Grernrum W, Beijersbergen RL, Bernards R (June 2016). "CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes". Nature Biotechnology. 34 (6): 631–3. doi:10.1038/nbt.3536. PMID 27111720.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (August 2012). "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity". Science. 337 (6096): 816–21. Bibcode:2012Sci...337..816J. doi:10.1126/science.1225829. PMC 6286148. PMID 22745249.
- Wu X, Kriz AJ, Sharp PA (June 2014). "Target specificity of the CRISPR-Cas9 system". Quantitative Biology. 2 (2): 59–70. doi:10.1007/s40484-014-0030-x. PMC 4338555. PMID 25722925.
- Zhang F, Wen Y, Guo X (September 2014). "CRISPR/Cas9 for genome editing: progress, implications and challenges". Human Molecular Genetics. 23 (R1): R40-6. doi:10.1093/hmg/ddu125. PMID 24651067.
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, et al. (April 2017). "Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening". Nature Protocols. 12 (4): 828–863. doi:10.1038/nprot.2017.016. PMC 5526071. PMID 28333914.
- Endo M, Mikami M, Endo A, Kaya H, Itoh T, Nishimasu H, et al. (January 2019). "Genome editing in plants by engineered CRISPR-Cas9 recognizing NG PAM". Nature Plants. 5 (1): 14–17. doi:10.1038/s41477-018-0321-8. PMID 30531939.
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. (February 2016). "Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9". Nature Biotechnology. 34 (2): 184–191. doi:10.1038/nbt.3437. PMC 4744125. PMID 26780180.
- Cancellieri S, Canver MC, Bombieri N, Giugno R, Pinello L (November 2019). "CRISPRitz: rapid, high-throughput, and variant-aware in silico off-target site identification for CRISPR genome editing". Bioinformatics. 36 (7): 2001–2008. doi:10.1093/bioinformatics/btz867. PMC 7141852. PMID 31764961.
- Sanjana NE, Shalem O, Zhang F (August 2014). "Improved vectors and genome-wide libraries for CRISPR screening". Nature Methods. 11 (8): 783–784. doi:10.1038/nmeth.3047. PMC 4486245. PMID 25075903.
- "Pooled Libraries". Addgene.
- Xu CL, Ruan MZ, Mahajan VB, Tsang SH (January 2019). "Viral Delivery Systems for CRISPR". Viruses. 11 (1): 28. doi:10.3390/v11010028. PMC 6356701. PMID 30621179.
- Lino CA, Harper JC, Carney JP, Timlin JA (November 2018). "Delivering CRISPR: a review of the challenges and approaches". Drug Delivery. 25 (1): 1234–1257. doi:10.1080/10717544.2018.1474964. PMC 6058482. PMID 29801422.
- Wang T, Wei JJ, Sabatini DM, Lander ES (January 2014). "Genetic screens in human cells using the CRISPR-Cas9 system". Science. 343 (6166): 80–4. Bibcode:2014Sci...343...80W. doi:10.1126/science.1246981. PMC 3972032. PMID 24336569.
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. (January 2014). "Genome-scale CRISPR-Cas9 knockout screening in human cells". Science. 343 (6166): 84–87. Bibcode:2014Sci...343...84S. doi:10.1126/science.1247005. PMC 4089965. PMID 24336571.
- Yaniz-Galende E, Hajjar RJ (January 2014). "Stem cell and gene therapy for cardiac regeneration.". Cardiac Regeneration and Repair. Woodhead Publishing. pp. 347–379. doi:10.1533/9780857096708.4.347. ISBN 9780857096586.
- Pauwels K, Gijsbers R, Toelen J, Schambach A, Willard-Gallo K, Verheust C, et al. (December 2009). "State-of-the-art lentiviral vectors for research use: risk assessment and biosafety recommendations". Current Gene Therapy. 9 (6): 459–74. doi:10.2174/156652309790031120. PMID 20021330.
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L (November 1998). "A third-generation lentivirus vector with a conditional packaging system". Journal of Virology. 72 (11): 8463–71. doi:10.1128/JVI.72.11.8463-8471.1998. PMC 110254. PMID 9765382.
- "Lenti-X CRISPR/Cas9 System User Manual" (PDF). Takara Bio USA.
- Tiscornia G, Singer O, Verma IM (2006). "Production and purification of lentiviral vectors". Nature Protocols. 1 (1): 241–5. doi:10.1038/nprot.2006.37. PMID 17406239.
- Agrotis A, Ketteler R (2015). "A new age in functional genomics using CRISPR/Cas9 in arrayed library screening". Frontiers in Genetics. 6: 300. doi:10.3389/fgene.2015.00300. PMC 4585242. PMID 26442115.
- Yeung AT, Choi YH, Lee AH, Hale C, Ponstingl H, Pickard D, et al. (October 2019). "Salmonella Infection". mBio. 10 (5). doi:10.1128/mBio.02169-19. PMC 6786873. PMID 31594818.
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, et al. (December 2015). "High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities". Cell. 163 (6): 1515–26. doi:10.1016/j.cell.2015.11.015. PMID 26627737.
- Slesarev A, Viswanathan L, Tang Y, Borgschulte T, Achtien K, Razafsky D, et al. (March 2019). "CRISPR/CAS9 targeted CAPTURE of mammalian genomic regions for characterization by NGS". Scientific Reports. 9 (1): 3587. Bibcode:2019NatSR...9.3587S. doi:10.1038/s41598-019-39667-4. PMC 6401131. PMID 30837529.
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. (2014). "MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens". Genome Biology. 15 (12): 554. doi:10.1186/s13059-014-0554-4. PMC 4290824. PMID 25476604.
- Li W, Köster J, Xu H, Chen CH, Xiao T, Liu JS, et al. (December 2015). "Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR". Genome Biology. 16: 281. doi:10.1186/s13059-015-0843-6. PMC 4699372. PMID 26673418.
- Sharma S, Petsalaki E (March 2018). "Application of CRISPR-Cas9 Based Genome-Wide Screening Approaches to Study Cellular Signalling Mechanisms". International Journal of Molecular Sciences. 19 (4): 933. doi:10.3390/ijms19040933. PMC 5979383. PMID 29561791.
- Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, et al. (October 2016). "A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia". Cell Reports. 17 (4): 1193–1205. doi:10.1016/j.celrep.2016.09.079. PMC 5081405. PMID 27760321.
- Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, et al. (January 2018). "CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2". The Journal of Clinical Investigation. 128 (1): 446–462. doi:10.1172/JCI90793. PMC 5749506. PMID 29202477.
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, et al. (November 2015). "Identification and characterization of essential genes in the human genome". Science. 350 (6264): 1096–101. Bibcode:2015Sci...350.1096W. doi:10.1126/science.aac7041. PMC 4662922. PMID 26472758.
- Leary M, Heerboth S, Lapinska K, Sarkar S (December 2018). "Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy". Cancers. 10 (12): 483. doi:10.3390/cancers10120483. PMC 6315347. PMID 30518036.
- Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, et al. (April 2019). "Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition". Oncogene. 38 (14): 2451–2463. doi:10.1038/s41388-018-0606-4. PMC 6450769. PMID 30532030.
- Hinze L, Pfirrmann M, Karim S, Degar J, McGuckin C, Vinjamur D, et al. (April 2019). "Synthetic Lethality of Wnt Pathway Activation and Asparaginase in Drug-Resistant Acute Leukemias". Cancer Cell. 35 (4): 664–676.e7. doi:10.1016/j.ccell.2019.03.004. PMC 6541931. PMID 30991026.
- Li B, Clohisey SM, Chia BS, Wang B, Cui A, Eisenhaure T, et al. (January 2020). "Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection". Nature Communications. 11 (1): 164. Bibcode:2020NatCo..11..164L. doi:10.1038/s41467-019-13965-x. PMC 6952391. PMID 31919360.
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, et al. (July 2016). "Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens". Nature. 535 (7610): 159–63. Bibcode:2016Natur.535..159M. doi:10.1038/nature18631. PMC 4964798. PMID 27383987.
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM (July 2015). "An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis". Cell. 162 (3): 540–51. doi:10.1016/j.cell.2015.07.016. PMC 4522279. PMID 26232224.
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera M, Yusa K (March 2014). "Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library". Nature Biotechnology. 32 (3): 267–73. doi:10.1038/nbt.2800. PMID 24535568.
- Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, et al. (March 2015). "Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis". Cell. 160 (6): 1246–60. doi:10.1016/j.cell.2015.02.038. PMC 4380877. PMID 25748654.
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR (June 2015). "Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains". Nature Biotechnology. 33 (6): 661–7. doi:10.1038/nbt.3235. PMC 4529991. PMID 25961408.
- Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, et al. (July 2015). "A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death". Cell Reports. 12 (4): 673–83. doi:10.1016/j.celrep.2015.06.049. PMC 4559080. PMID 26190106.
- Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, et al. (July 2015). "A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks". Cell. 162 (3): 675–86. doi:10.1016/j.cell.2015.06.059. PMC 4522370. PMID 26189680.
- Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V (January 2016). "A Genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation". The Journal of Biological Chemistry. 291 (1): 103–9. doi:10.1074/jbc.C115.700492. PMC 4697147. PMID 26553871.
- Quinn T. "Advantages and challenges of pooled libraries". Webinar series. Takara Bio USA.
- Fang Z, Weng C, Li H, Tao R, Mai W, Liu X, et al. (March 2019). "Single-Cell Heterogeneity Analysis and CRISPR Screen Identify Key β-Cell-Specific Disease Genes". Cell Reports. 26 (11): 3132–3144.e7. doi:10.1016/j.celrep.2019.02.043. PMC 6573026. PMID 30865899.
- Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M (January 2013). "Viral vectors: a look back and ahead on gene transfer technology". The New Microbiologica. 36 (1): 1–22. PMID 23435812.
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, et al. (December 2016). "Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq". Cell. 167 (7): 1883–1896.e15. doi:10.1016/j.cell.2016.11.039. PMID 27984734.
- Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, et al. (March 2017). "Pooled CRISPR screening with single-cell transcriptome readout". Nature Methods. 14 (3): 297–301. doi:10.1038/nmeth.4177. PMC 5334791. PMID 28099430.
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. (December 2016). "Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens". Cell. 167 (7): 1853–1866.e17. doi:10.1016/j.cell.2016.11.038. PMC 5181115. PMID 27984732.