Epitranscriptomic sequencing
In epitranscriptomic sequencing, most methods focus on either (1) enrichment and purification of the modified RNA molecules before running on the RNA sequencer, or (2) improving or modifying bioinformatics analysis pipelines to call the modification peaks. Most methods have been adapted and optimized for mRNA molecules, except for modified bisulfite sequencing for profiling 5-methylcytidine which was optimized for tRNAs and rRNAs.
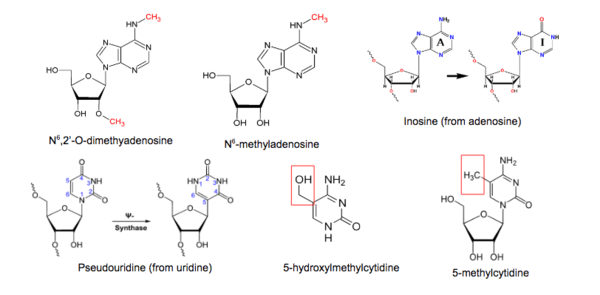
There are six major classes of chemical modifications found in RNA molecules: N6-methyladenosine, N6,2'-O-dimethyladenosine, 5-methylcytidine, 5-hydroxylmethylcytidine, inosine, and pseudouridine.[1][2] Various sequencing methods have been developed to profile each type of modification. The scale, resolution, sensitivity, and limitations associated with each method and the corresponding bioinformatics tools used will be discussed.
Methods for profiling N6-methyladenosine
Methylation of adenosine does not affect its ability to base-pair with thymidine or uracil, so N6-methyladenosine (m6A) cannot be detected using standard sequencing or hybridization methods.[3] This modification is marked by the methylation of the adenosine base at the nitrogen-6 position. It is abundantly found in polyA+ mRNA; also found in tRNA, rRNA, snRNA, and long ncRNA.
m6A-seq and MeRIP-seq
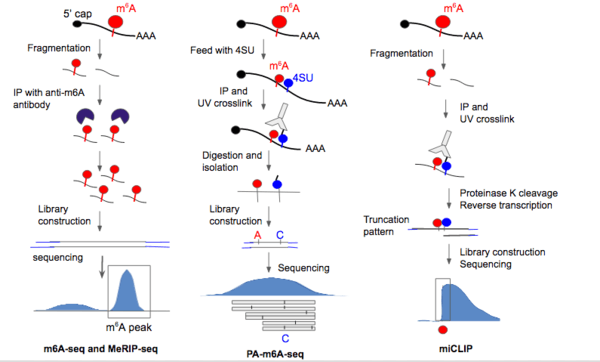
In 2012, the first two methods for m6A sequencing came out that enabled transcriptome-wide profile of m6A in mammalian cells.[1] These two techniques, called m6A-seq and MeRIP-seq (m6A-specific methylated RNA immunoprecipitation), are also the first methods to allow for any type of RNA modification sequencing. These methods were able to detect 10,000 m6A peaks in the mammalian transcriptome; the peaks were found to be enriched in 3’UTR regions, near STOP codons, and within long exons.[4][3]
The two methods were optimized to detect methylation peaks in poly(A)+ mRNA, but the protocol could be adapted to profile any type of RNA. Collected RNA sample is fragmented into ~100-nucleotide-long oligonucleotides using a fragmentation buffer, immunoprecipitation with purified anti-m6A antibody, elution and collection of antibody-tagged RNA molecules. The immunoprecipitation procedure in MeRIP-Seq is able to produce >130fold enrichment of m6A sequences.[3] Random primed cDNA library generation was performed, followed by adaptor ligation and Illumina sequencing. Since the RNA strands are randomly chopped up, the m6A site should, in principle, lie somewhere in the center of the regions to which sequence reads align. At extremes, the region would be roughly 200nt wide (100nt up- and downstream of the m6A site).[3]
When the first nucleotide of a transcript is an adenosine, in addition to the ribose 2’-O-methylation, this base can be further methylated at the N6 position.[1] m6A-seq was confirmed to be able to detect m6Am peaks at transcription start sites.[5][4] Adapter ligation at both ends of RNA fragment results in reads tending to pileup at the 5’ terminus of the transcript. Schwartz et al. (2015) leveraged this knowledge to detect mTSS sites by picking out sites with a high ratio of the size of pileups in the IP samples compared to input sample.[4] As confirmation, >80% of the highly enriched pileup sites contained adenosine.
The resolution of these methods is 100-200nt, which was the range of the fragment size. These two methods had several drawbacks: (1) required substantial input material, (2) low resolution which made pinpointing the actual site with the m6A mark difficult, and (3) cannot directly assess false positives.[6]
Especially in MeRIP-Seq, the bioinformatics tools that are currently available are only able to call 1 site per ~100-200nt wide peak, so a substantial portion of clustered m6As (~64nt between each individual site within a cluster) are missed.[7][8] Each cluster can contain up to 15 m6A residues.[7][8]
In 2013, a modified version of m6A-seq based on the previous two methods m6A-seq and MeRIP-seq came out which aimed to increase resolution, and demonstrated this in the yeast transcriptome. They achieved this by decreasing fragment size and employing a ligation-based strand-specific library preparation protocol capturing both ends of the fragmented RNA, ensuring that the methylated position is within the sequenced fragment.[6] By additionally referencing the m6A consensus motif and eliminating false positive m6A peaks using negative control samples, the m6A profiling in yeast was able to be done at single-base resolution.
UV-based Methods
PA-m6A-seq
UV-induced RNA-antibody crosslinking was added on top of m6A-seq to produce PA-m6A-seq (photo-crosslinking-assisted m6A-seq) which increases resolution up to ~23nt. First, 4-thiourodine (4SU) is incorporated into the RNA by adding 4SU in growth media, some incorporation sites presumably near m6A location. Immunoprecipitation is then performed on full-length RNA using m6A-specific antibody [36]. UV light at 365 nm is then shined onto RNA to activate the crosslinking to the antibody with 4SU. Crosslinked RNA was isolated via competition elution and fragmented further to ~25-30nt; proteinase K was used to dissociate the covalent bond between crosslinking site and antibody.[6] Peptide fragments that remain after antibody removal from RNA cause the base to be read as a C as opposed to a T during reverse transcription, effectively inducing a point mutation at the 4SU crosslinking site.[6] The short fragments are subjected to library construction and Illumina sequencing, followed by finding the consensus methylation sequence. The presence of the T to C mutation helps increase the signal to noise ratio of methylation site detection [1] as well as providing greater resolution to the methylation sequence. One shortcoming of this method is that m6A sites that did not incorporate 4SU can’t be detected. Another caveat is that position of 4SU incorporation can vary relative to any single m6A residue, so it still remains challenging to precisely locate m6A site using the T to C mutation.
m6A-CLIP and miCLIP
m6A-CLIP [7] (crosslinking immunoprecipitation) and miCLIP [8] (m6A individual-nucleotide-resolution crosslinking and immunoprecipitation) are UV-based sequencing techniques. These two methods activate crosslinking at 254 nm, fragments RNA molecules before immunoprecipitation with antibody, and do not depend on the incorporation of photoactivatable ribonucleosides - the antibody directly crosslinks with a base close (very predictable location) to the m6A site. These UV-based strategies uses antibodies that induces consistent and predictable mutational and truncation patterns in the cDNA strand during reverse-transcription that could be leveraged to more precisely locate the m6A site.[7][8] Though both m6A-CLIP and miCLIP reply on UV induced mutations, m6A-CLIP [7] is distinct by taking advantage that m6A alone can induce cDNA truncation during reverse transcription and generate single-nucleotide mapping for over ten folds more precise m6A sites (MITS, m6A-induced truncation sites), permitting comprehensive and unbiased precise m6A mapping.[7] In contrast, UV-mapped m6A sites by miCLIP is only a small subset of total precise m6A sites. The precise location of tens of thousands of m6A sites in human and mouse mRNAs by m6A-CLIP reveals that m6A is enriched at last exon but not around stop codon.[7]
In m6A-CLIP [7] and miCLIP,[8] RNA is fragmented to ~20-80nt first, then the 254 nm UV-induced covalent RNA/m6A antibody complex was formed in the fragments containing m6A. The antibody was removed with proteinase K before reverse-transcription, library construction and sequencing. Remnants of peptides at the crosslinking site on the RNA after antibody removal, leads to insertions, truncations, and C to T mutations during reverse transcription to cDNA, especially at the +1 position to the m6A site (5’ to the m6A site) in the sequence reads.[7][8] Positive sites seen using m6A-CLIP and miCLIP had high percent of matches with those detected using SCARLET, which has higher local resolution around a specific site, (see below), implicating m6A-CLIP and miCLIP has high spatial resolution and low false discovery rate.[7][8] miCLIP has been used to detect m6Am by looking at crosslinking-induced truncation sites at the 5’UTR.[8]
Methods for quantifying m6A modification status
Although m6A sites could be profiled at high resolution using UV-based methods, the stoichiometry of m6A sites - the methylation status or the ratio m6A+ to m6A- for each individual site within a type of RNA - is still unknown. SCARLET (2013) and m6A-LAIC-seq (2016) allows for the quantitation of stoichiometry at a specific locus and transcriptome-wide, respectively.[1]
Bioinformatics methods used to analyze m6A peaks do not make any prior assumptions about the sequence motifs within which m6A sites are usually found, and take into consideration all possible motifs. Therefore, it is less likely to miss sites.[8]
SCARLET
SCARLET (site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography) is used determining the fraction of RNA in a sample that carries a methylated adenine at a specific site. One can start with total RNA without having to enrich for the target RNA molecule. Therefore, it is an especially suitable method for quantifying methylation status in low abundance RNAs such as tRNAs. However, it is not suitable or practical for large-scale location of m6A sites.[8][9]
The procedure begins with a chimeric DNA oligonucleotide annealing to the target RNA around the candidate modification site. The chimeric ssDNA has 2’OMe/2’H modifications and is complementary to the target sequence. The chimeric oligonucleotide serves as a guide to allow RNase H to cleave the RNA strand precisely at the 5’-end of the candidate site. The cut site is then radiolabeled with phosphorus-32 and splint-ligated to a 116nt ssDNA oligonucleotide using DNA ligase.[10] RNase T1/A is introduced to the sample to digest all RNA, except for the RNA molecules with the 116-mers DNA attached. This radiolabeled product is then isolated and digested by nuclease to generate a mixture of modified and unmodified adenosines (5’P-m6A and 5’-P-A) which is separated using thin layer chromatography. The relative proportions of the two groups can be determined using UV absorption levels.[9]
m6A-LAIC-seq
m6A-LAIC-seq (m6A-level and isoform-characterization sequencing) is a high-throughput approach to quantify methylation status on a whole-transcriptome scale. Full-length RNA samples are used in this method. RNAs are first subjected to immunoprecipitation with an anti-m6A antibody. Excess antibody is added to the mixture to ensure all m6A-containing RNAs are pulled down. The mixture is separated into eluate (m6A+ RNAs) and supernatant (m6A- RNAs) pools. External RNA Controls Consortium (ERCC) spike ins are added to the eluate and supernatant, as well as an independent control arm consisting of just ERCC spike in. After antibody cleavage in the eluate pool, each of the three mixtures are sequenced on a next generation sequencing platform. The m6A levels per site or gene could be quantified by the ERCC-normalized RNA abundances in different pools.[1][11] Since full-length RNA is used, it is possible to directly compare alternatively spliced isoforms between the m6A+ and m6A- fractions as well as comparing isoform abundance within the m6A+ portion.
Despite the advances in m6A-sequencing, several challenges still remain: (1) A method has yet to be developed that characterizes the stoichiometry between different sites in the same transcript; (2) Analysis results are heavily dependent on the bioinformatics algorithm used to call the peaks; (3) Current methods all use m6A-specific antibodies to tag m6A sites, but it has been reported that the antibodies contain intrinsic bias for RNA sequences.
Methods for N6,2'-O-dimethyladenosine (m6Am) Profiling
N6,2'-O-dimethyladenosine, abundant in polyA+ mRNAs, occurs at the first nucleotide after the 5’ cap, when an additional methyl group is added to a 2ʹ-O-methyladenosine residue at the ‘capped’ 5ʹ end of mRNA.[1]
Since m6Am can be recognized by anti-m6A antibodies at transcription start sites, the methods used for m6A profiling can be and were adapted for m6Am profiling, namely m6A-seq, and miCLIP (see m6A-seq and miCLIP descriptions above).
Methods for 5-methylcytidine profiling
5-methylcytidine, m5C, is abundantly found in mRNA and ncRNAs, especially tRNA and rRNAs. In tRNAs, this modification stabilizes the secondary structure and influences anticodon stem-loop conformation.[1] In rRNAs, m5C affects translational fidelity.[1]
Two principles have been used to develop m5C sequencing methods. The first one is antibody-based approach (bisuphite sequencing and m5C-RIP), similar to m6C sequencing. The second is detecting targets of m5C RNA methyltransferases by covalently linking the enzyme to its target, and then using IP specific to the target enzyme to enrich for RNA molecules containing the mark (Aza-IP and miCLIP).
Modified bisulfite sequencing
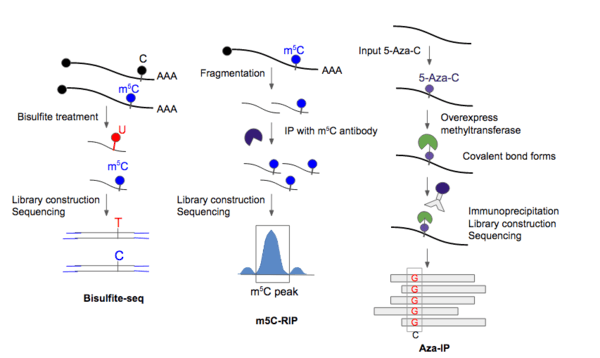
Modified bisulfite sequencing was optimized for rRNA, tRNA, and miRNA molecules from Drosophila.[12] Bisulfite treatment has been most widely used to detect dm5C (DNA m5C). The treatment essentially converts a cytosine to a uridine, but methylated cytosines would be unchanged by the treatment. Previous attempts to develop m5C sequencing protocols using bisulfite treatment were not able to effectively address the problem of the harsh treatment of RNA which causes significant degradation of the molecules. Specifically, bisulfite deamination treatment (high pH) of RNA is detrimental to the stability of phosphodiester bonds. As a result, it is difficult to pre-enrich RNA molecules or to obtain enough PCR product of the correct size for deep sequencing.
A modified version of bisulfite sequencing was developed by Schaefer et al. (2009) which decreased the temperature at which bisulfite treatment of RNA from 95 °C to 60 °C.[12] The rationale behind the modification was that since RNA, unlike DNA, is not double-stranded, but rather, consists of regions of single-strandedness, double-stranded stem structures and loops, it could be possible to unwind RNA at a much lower temperature. Indeed, RNA could be treated for 180 minutes at 60C without significant loss of PCR amplicons of the expected size.[12] Deamination rates were determined to be 99% at 180min of treatment.
After bisulfite treatment of fragmented RNA, reverse transcription is performed, followed by PCR amplification of the cDNA products, and finally deep sequencing was done using the Roche 454 platform.[12]
Since the developers of the method used the Roche platform, they also used GS Amplicon Variant Analyzer (Roche) for analyzing deep sequencing data to quantify sequence-specific cytosine content. However, recent papers have suggested that the method have several flaws: (1) Incomplete conversion of regular cytosines in double-stranded regions of RNA; (2) areas containing other modifications that resulted in bisulfite-treatment resistance; and (3) sites containing potential false-positives due to (1) and (2) [13][14][15] In addition, it is possible the sequencing depth is still not high enough to correctly detect all methylated sites.[1]
Aza-IP
Aza-IP 5-azacytidine-mediated RNA immunoprecipitation has been optimized on and used for detecting targets of methyltransferases, particularly NSUN2 and DNMT2 [16] — the two main enzymes responsible for laying down the m5C mark.
First, the cell is made to overexpress an epitope-tagged m5C-RNA methytransferase derivative so that the antibody used later on for immunoprecipitation could recognize the enzyme. Second, 5-aza-C is introduced to the cells so that it could be incorporated into nascent RNA in place of cytosine. Normally, the methyltransferases are released (i.e. covalent bond between cytosine and methyltransferase is broken) following methylation of the residue. For 5-aza-C, due to a nitrogen substitution in the C5 position of cytosine, the RNA methytransferase enzyme remains covalently bound to the target RNA molecule at the C6 position.[16]
Third, the cell is lysed and the m5C-RNA methyltransferase of interest is immunoprecipitated along with the RNA molecules that are covalently linked to the protein. The IP step enabled >200-fold enrichment of RNA targets, which were mainly tRNAs. The enriched molecules were then fragmented and purified. cDNA library is then constructed and sequencing is performed.[16]
An important additional feature is that RNA methyltransferase covalent linkage to the C5 of m-aza-C induces rearrangement and ring opening. This ring opening results in preferential pairing with cytosine and is therefore read as guanosine during sequencing. This C to G transversion allows for base resolution detection of m5C sites.[1] One caveat is that m5C sites not replaced by 5-azacytosine will be missed.
miCLIP
miCLIP (Methylation induced crosslinking immunoprecipitation) was used to detect NSUN2 targets, which were found to be mostly non-coding RNAs such as tRNA. An induced mutation of C271A in NSUN2 inhibits release of enzyme from RNA target. This mutation was over-expressed in the cells of interest, and the mutated NSUN2 was also tagged with the Myc epitope. The covalently linked RNA-protein complexes are isolated via immunoprecipitation for a Myc-specific antibody. These complexes are confirmed and detected by radiolabeling with phosphorus-32. The RNA is then extracted from the complex, reverse-transcribed, amplified with PCR, and sequenced using next-generation platforms.
Both miCLIP and Aza-IP, though limited by specific targeting of enzymes, can allow for the detection of low-abundance methylated RNA without deep sequencing.[1]
Methods for Inosine Profiling
Inosine is created enzymatically when an adenosine residue is modified.
Analysis of base-pairing properties
Since the chemical makeup of inosine is a deaminated adenosine, this is one of few methylation alterations that has an accompanying alteration in base pairing, which can be capitalised on. The original adenosine nucleotide will pair with a thymine, whereas the methylated inosine will pair with a cytosine. cDNA sequences obtained by rtPCR can therefore be compared to the corresponding genomic sequences; in sites where A residues are repeatedly interpreted as G, a methylation event can be assumed. At high enough accuracy, it is feasible that the quantity of mRNA molecules in the population that have been methylated can be calculated as a percentage. This method potentially has single-nucleotide resolution. In fact, the abundance of RNA-seq data that is now publicly available can be leveraged to investigate G (in cDNA) versus A (in genome). One particular pipeline, called RNA and DNA differences (RDD), claims to excludes false positives, but only 56.8% of its A-to-I sites were found to be valid by ICE-seq[17](see below).
Limitations
The background noise caused by single nucleotide polymorphisms (SNPs), somatic mutations, pseudogenes and sequencing errors reduce the reliability of the signal, especially in a single-cell context.[18]
Chemical methods
Inosine-specific cleavage
The first method to detect A-to-I RNA modifications, developed in 1997, was inosine-specific cleavage. RNA samples are treated with glyoxal and borate to specifically modify all G bases, and subsequently enzymatically digested to by RNase T1, which cleaves after I sites. The amplification of these fragments then allows analysis of cleavage sites and inference of A-to-I modification. .[19] It was used to prove the position of inosine at specific sites rather than identify novel sites or transcriptome-wide profiles.
Limitations
The existence of two A-to-I modifications in relatively close proximity, which is common in Alu elements, means the downstream mod is less likely to be detected since the cDNA synthesis will be truncated at a prior nucleotide. The throughput is low, and the initial method required specific primers; the protocol is complicated and labour-intensive.
ICE and ICE-seq
Inosine chemical erasing (ICE) refer to a process in which acrylonitrile is reacted with inosine to form N1-cyanoethylinosine (ce1I). This serves to stall reverse transcriptase and lead to truncated cDNA molecules. This was combined with deep-sequencing in a developed method called ICE-seq. Computational methods for automated analysis of the data are available, the main premise being the comparison of treated and untreated samples to identify truncated transcripts and thus infer an inosine modification by read count, with a step to reduce false positives by comparison to online database dbSNP.[20]
Limitations
The original ICE protocol involved an RT-PCR amplification step and therefore required primers and knowledge of the location or regions to be investigated,[21] alongside a maximum cDNA length of 300–500bp. The ICE-seq method is complicated, along with being labour-, reagent- and time-intensive. One protocol from 2015 took 22 days.[21][20][17] This shares a limitation with inosine-specific cleavage, in that if there are two A-to-I modifications in relatively close proximity, the downstream mod is less likely to be detected since the cDNA synthesis will be truncated at a prior nucleotide.[22] Both ICE and ICE-seq suffer from a lack of sensitivity to infrequently edited locations: it becomes difficult to distinguish a modification with a frequency of <10% from a false positive. An increase in read depth and quality can increase sensitivity, but also then suffer from further amplification bias.
Biological methods
ADAR knockdown
The modification of A to I is effected by adenosine deaminases that act on RNA (ADARs), of which in mice there are three. The knockdown of these in the cell, therefore, and the subsequent cell–cell comparison of ADAR+ and ADAR- RNA content would be anticipated to provide a basis for A-to-I modification profiling. However, there are further functions of ADAR enzymes within the cell — for example, they have further roles in RNA processing, and in miRNA biogenesis — which would also be likely to change the landscape of cellular mRNA. Recently a map of A-to-I editing in mice was generated using editing-deficient ADAR1 and ADAR2 double-knockout mice as a negative control. Thereby, A-to-I editing was detected with high confidence.[23]
Methods for Pseudouridine Methylation Profiling
Pseudouridine, or Ψ, the overall most abundant post-translational RNA modification,[24] is created when a uridine base is isomerised. In eukaryotes, this can occur by either of two distinct mechanisms;[25][26] it is sometimes referred to as the ‘fifth RNA nucleotide’. It is incorporated into stable non-coding RNAs such as tRNA, rRNA, and snRNA,[27] with roles in ribosomal ligand binding and translational fidelity in tRNA,[28][29] and in fine-tuning branching events and splicing events in snRNAs.[30] Pseudouridine has one more hydrogen bond donor from an imino group and a more stable C–C bond, since a C-glycosidic linkage has replaced the N-glycosidic linkage found in its counterpart (regular uridine).[31] As neither of these changes affect its base-pairing properties, both will have the same output when directly sequenced; therefore methods for its detection involve prior biochemical modification.[32]
Biochemical methods
CMCT methods
There are multiple pseudouridine detection methods beginning with the addition of N-cyclohexyl-N′-b-(4-methylmorpholinium) ethylcarbodiimide metho-p-toluene-sulfonate (CMCT; also known as CMC), since its reaction with pseudouridine produces CMC-Ψ. CMC-Ψ causes reverse transcriptase to stall one nucleotide in the 3’ direction.[33] These methods have single-nucleotide resolution. In an optimisation step, azido-CMC can confer the ability to add biotinylation; subsequent biotin pulldown will enrich Ψ-containing transcripts, allowing identification of even low-abundance transcripts.
Limitations
As with other procedures predicated on biochemical alteration followed by sequencing, the development of high-throughput sequencing has removed the limitations requiring prior knowledge of sites of interest and primer design. The method causes a lot of RNA degradation, so it is necessary to start with a large amount of sample, or use effective normalisation techniques to account for amplification biases. One final limitation is that, for CMC labelling of pseudouridine to be specific, it is not complete, and therefore nor is it quantitative.[34] A new reactant that could achieve a higher sensitivity with specificity would be beneficial.
Methods for 5-hydroxylmethylcytidine Profiling
Cytidine residues, modified once to m5C (discussed above), can be further modified: either oxidised once for 5-hydroxylmethylcytidine (hm5C), or oxidised twice for 5-formylcytidine (f5C). Arising from the oxidative processing of m5C enacted in mammals by ten-eleven translocation (TET) family enzymes, hm5C is known to occur in all three kingdoms and to have roles in regulation. While 5-hydroxymethylcytidine (hm5dC) is known to be found in DNA in a widespread manner, hm5C is also found in organisms for which no hm5dC has been detected, indicating it is a separate process with distinct regulatory stipulations. To observe the in vivo addition of methyl groups to cytosine RNA residues followed by oxidative processing, mice can be fed on a diet incorporating particular isotopes and these be traced by LC-MS/MS analysis. Since the metabolic pathway from nutritional intake to nucleotide incorporation is known to progress from dietary methionine --> S-adenosylmethionine (SAM) --> methyl group on RNA base, the labelling of dietary methionine with 13C and D means these will end up in hm5C residues that have been altered since the addition of these into the diet.[35] In contrast to m5C, a large quantity of hm5C modifications have been recorded within coding sequences.[36]
hMeRIP-seq
hMeRIP-seq is an immunoprecipitation method, in which RNA–protein complexes are crosslinked for stability, and antibodies specific to hm5C are added. Using this method, over 3,000 hm5C peaks have been called in Drosophila melanogaster S2 cells.
Limitations
Despite two distinct base-resolution methods being available for hm5dC, there are no base-resolution methods for detection of hm5C.
Biophysical validation of RNA modifications
Apart from mass spectrometry and chromatography, other two validation techniques have been developed, namely
- Pre- and post-labelling techniques:
- Pre-labelling → involves the use of 32P: cells are grown in 32P containing medium, thus allowing the incorporation of [α-32P]NTPs during transcription by T7 RNA polymerase. The modified RNA is then extracted, and each RNA species is isolated and subsequently digested by T2 RNase. Next, RNA is hydrolyzed into 5' nucleoside monophosphates, which are analyzed 2D-TLC (two-dimensional thin-layer chromatography). This method is able to detect and quantify every modification but will not contribute to the characterization of the sequence.
- Post-labelling → implicates the selective labelling of a specific position within the sequence: these techniques rely on the Stanley-Vassilenko approach principles, that has been adjusted to achieve a better validation quality. First, RNA is cleaved into free 5’-OH fragments either by RNase H or DNAzymes, by sequence specific hydrolysis. The polynucleotide kinase (PKN) then performs the 5’ radioactive post-labelling phosphorylation using [γ-32P]ATP. At this point, the labelled fragments undergo a size fragmentation, that can be performed either by Nuclease P1 or according to the SCARLET method. In both cases, the final product is a group of 5’ nucleoside monophosphates (5’ NMPs) that will be analyzed by TLC.
- SCARLET: this recent approach exploits not just one, but two sequence selection steps, the last of which is obtained during the splinted ligation of the radioactive-labelled fragments with a long DNA oligonucleotide, at its 3’-end. After degradation, the labelled residue is purified together with the ligated DNA oligonucleotide and finally hydrolyzed and therefore released thanks to the activity of the Nuclease P1.
This method has proven to be very useful in the validation of modified residues in mRNAs and lncRNAs, such as m6A and Ψ
- Oligonucleotide-based techniques: this method includes several variants
- Splinted ligation of particular modified DNAs, that exploits the ligase sensitivity to 3’ and 5’ nucleotides (so far used for m6A, 2’-O-Me, Ψ)
- Microarray modification identification through a DNA-chip, that exploits the decrease in duplex stability of cDNA oligonucleotides, due to the impediment in conventional base-pairing caused by modifications (ex. m1A, m1G, m22G)
- RT primer extension at low dNTPs concentration, for mapping of RT arrest signals.[36]
Single-Molecule Real-Time Sequencing for epitranscriptome sequencing
Single-molecule real-time sequencing (SMRT) is used in the epigenomic and epitranscriptomic fields. As regards epigenomics, thousands of zero-mode waveguides (ZMWs) are used to capture the DNA polymerase: when a modified base is present, the biophysical dynamics of its movement changes, creating a unique kinetic signature before, during, and after the base incorporation. SMRT sequencing can be used to detect modified bases in RNA, including m6A sites. In this case, a reverse transcriptase is used as enzyme with ZMWs to observe the cDNA synthesis in real time. The incorporation of synthetically designed m6A sites leaves a kinetic signature and increases the interpulse duration (IPD). There are some issues concerning the reading of homonucleotide straches and the base resolution of m6A therein, due to the stuttering of reverse transcriptase. Secondly, the throughput is too low for transcriptome-wide approaches. One of the most commonly used platform is the SMRT sequencing technology by Pacific Biosciences.[37]
Nanopore sequencing in epitranscriptomics
A possible alternative to the detection of epitranscriptomic modifications by SMRT sequencing is the direct detection using the Nanopore sequencing technologies. This technique exploits nanometer-sized protein channels embedded into a membrane or solid materials, and coupled to sensors, able to detect the amplitude and duration of the variations of the ionic current passing through the pore. As the RNA passes through the nanopore, the blockage leads to a disruption in current stream, which is different for the different bases, included modified ones, and therefore can be used to identify possible modifications. By producing single-molecule reads, without previous RNA amplification and conversion to cDNA, these techniques can lead to the production of quantitative transcriptome-wide maps.[1] In particular, the Nanopore technology proved to be effective in detecting the presence of two nucleotide analogs in RNA: N6-methyladenosine (m6A) and 5-methylcytosine (5-mC). Using Hidden Markov Models (HMM) or recurrent neural networks (RNN) trained with known sequences, it was possible to demonstrate that the modified nucleotides produce a characteristic disruption in the ionic current when passing through the pore, and that these data can be used to identify the nucleotide.[1][38]
References
- Li, X, Xiong, X., and Yi, C. (2017). Epitranscriptomic Sequencing Technologies: decoding RNA modifications. Nature Methods. doi:10.1038/NMETH_4110
- Licht, Konstantin; Jantsch, Michael F. (2016-04-11). "Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications". Journal of Cell Biology. 213 (1): 15–22. doi:10.1083/jcb.201511041. ISSN 0021-9525. PMC 4828693. PMID 27044895.
- Meyer, K.D.; et al. (2012). "Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3'UTRs and near Stop Codons". Cell. 149 (7): 1635–1646. doi:10.1016/j.cell.2012.05.003. PMC 3383396. PMID 22608085.
- Dominissini; et al. (2012). "Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq". Nature. 485 (7397): 201–208. Bibcode:2012Natur.485..201D. doi:10.1038/nature11112. PMID 22575960.
- Schwartz, S. et al. (2014). Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 50 Sites. Cell Reports. 8: 284-296.
- Schwartz, S.; et al. (2013). "High-Resolution Mapping Reveals a Conserved, Widespread, Dynamic mRNA Methylation Program in Yeast Meiosis". Cell. 155 (6): 1409–1421. doi:10.1016/j.cell.2013.10.047. PMC 3956118. PMID 24269006.
- Ke, S.; et al. (2015). "A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation". Genes & Development. 29 (19): 2037–2053. doi:10.1101/gad.269415.115. PMC 4604345. PMID 26404942.
- Linder, B.; et al. (2015). "Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome". Nature Methods. 12 (8): 767–772. doi:10.1038/nmeth.3453. PMC 4487409. PMID 26121403.
- Maity, A.; Das, B. (2016). "N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases". FEBS Journal. 283 (9): 1607–1630. doi:10.1111/febs.13614. PMID 26645578.
- Liu; et al. (2013). "Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA". RNA. 19 (12): 1848–1856. doi:10.1261/rna.041178.113. PMC 3884656. PMID 24141618.
- Molinie, B.; et al. (2016). "m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome". Nature Methods. 13 (8): 692–698. doi:10.1038/nmeth.3898. PMC 5704921. PMID 27376769.
- Schaefer, M.; et al. (2008). "RNA cytosine methylation analysis by bisulphite sequencing". Nucleic Acids Research. 37 (2): e12. doi:10.1093/nar/gkn954. PMC 2632927. PMID 19059995.
- Gilbert, W.V.; Bell, T.A.; Schaening, C. (2016). "Messenger RNA modifications: form, distribution, and function". Science. 352 (6292): 1408–1412. Bibcode:2016Sci...352.1408G. doi:10.1126/science.aad8711. PMC 5094196. PMID 27313037.
- Hussain, S.; Aleksic, J.; Blanco, S.; Dietmann, S.; Frye, M. (2013). "Characterizing 5-methylcytosine in the mammalian epitranscriptome". Genome Biol. 14 (11): 215. doi:10.1186/gb4143. PMC 4053770. PMID 24286375.
- Shafik, A.; Schumann, U.; Evers, M.; Sibbritt, T.; Preiss, T. (2016). "The emerging epitranscriptomics of long noncoding RNAs". Biochim. Biophys. Acta. 1859 (1): 59–70. doi:10.1016/j.bbagrm.2015.10.019. PMID 26541084.
- Khoddami, V. and Cairns, B.R. (2013). Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nature Biotechnology. 31(5): 459-464.
- Sakurai, M.; et al. (2014). "A biochemical landscape of A-to-I RNA editing in the human brain transcriptome". Genome Res. 24 (3): 522–534. doi:10.1101/gr.162537.113. PMC 3941116. PMID 24407955.
- Athanasiadis, Alekos; Rich, Alexander; Maas, Stefan (2004). "Widespread A-to-I RNA Editing of Alu-Containing mRNAs in the Human Transcriptome". PLOS Biology. 2 (12): e391. doi:10.1371/journal.pbio.0020391. PMC 526178. PMID 15534692.
- Morse, D.P.; Bass, B.L. (1997). "Detection of inosine in messenger RNA by inosine-specific cleavage". Biochemistry. 36 (28): 8429–8434. doi:10.1021/bi9709607. PMID 9264612.
- Sakurai, Masayuki; Yano, Takanori; Kawabata, Hitomi; Ueda, Hiroki; Suzuki, Tsutomu (2010). "Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome". Nature Chemical Biology. 6 (10): 733–740. doi:10.1038/nchembio.434. PMID 20835228.
- Morse, D. P.; Bass, B. L. (1997). "Detection of inosine in messenger RNA by inosine-specific cleavage". Biochemistry. 36 (28): 8429–8434. doi:10.1021/bi9709607. PMID 9264612.
- Suzuki, Tsutomu; Ueda, Hiroki; Okada, Shunpei; Sakurai, Masayuki (2015). "Transcriptome-wide identification of adenosine-to-inosine editing using the ICE-seq method". Nature Protocols. 10 (5): 715–732. doi:10.1038/nprot.2015.037. PMID 25855956.
- Licht, Konstantin; Kapoor, Utkarsh; Amman, Fabian; Picardi, Ernesto; Martin, David; Bajad, Prajakta; Jantsch, Michael F. (September 2019). "A high resolution A-to-I editing map in the mouse identifies editing events controlled by pre-mRNA splicing". Genome Research. 29 (9): 1453–1463. doi:10.1101/gr.242636.118. ISSN 1088-9051. PMC 6724681. PMID 31427386.
- Charette, M.; Gray, M.W. (2000). "Pseudouridine in RNA: what, where, how, and why". IUBMB Life. 49 (5): 341–351. doi:10.1080/152165400410182. PMID 10902565.
- Kiss, T.; Fayet-Lebaron, E.; Jady, B.E. (2010). "Box H/ACA small ribonucleoproteins". Mol. Cell. 37 (5): 597–606. doi:10.1016/j.molcel.2010.01.032. PMID 20227365.
- Hamma, T.; Ferre-D; Amare, A.R. (2006). "Pseudouridine synthases". Chem. Biol. 13 (11): 1125–1135. doi:10.1016/j.chembiol.2006.09.009. PMID 17113994.
- Karijolich, J.; Yi, C.; Yu, Y.T. (2015). "Transcriptome-wide dynamics of RNA pseudouridylation". Nat. Rev. Mol. Cell Biol. 16 (10): 581–585. doi:10.1038/nrm4040. PMC 5694666. PMID 26285676.
- Jack, K.; et al. (2011). "rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells". Mol. Cell. 44 (4): 660–666. doi:10.1016/j.molcel.2011.09.017. PMC 3222873. PMID 22099312.
- Baudin-Baillieu, A.; et al. (2009). "Nucleotide modifications in three functionally important regions of the Saccharomyces cerevisiae ribosome affect translation accuracy". Nucleic Acids Res. 37 (22): 7665–7677. doi:10.1093/nar/gkp816. PMC 2794176. PMID 19820108.
- Yu, A.T.; Ge, J.; Yu, Y.T. (2011). "Pseudouridines in spliceosomal snRNAs". Protein Cell. 2 (9): 712–725. doi:10.1007/s13238-011-1087-1. PMC 4722041. PMID 21976061.
- Basturea, Georgeta N. (2013). "Research Methods for Detection and Quantitation of RNA Modifications". Materials and Methods. 3. doi:10.13070/mm.en.3.186.
- Carlile, Thomas M.; Rojas-Duran, Maria F.; Zinshteyn, Boris; Shin, Hakyung; Bartoli, Kristen M.; Gilbert, Wendy V. (2014). "Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells". Nature. 515 (7525): 143–146. Bibcode:2014Natur.515..143C. doi:10.1038/nature13802. PMC 4224642. PMID 25192136.
- Bakin, A.; Ofengand, J. (1993). "Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: analysis by the application of a new sequencing technique". Biochemistry. 32 (37): 9754–9762. doi:10.1021/bi00088a030. PMID 8373778.
- Kellner, S.; Burhenne, J.; Helm, M. (2010). "Detection of RNA modifications". RNA Biol. 7 (2): 237–247. doi:10.4161/rna.7.2.11468. PMID 20224293.
- Huber, Sabrina M.; van Delft, Pieter; Mendil, Lee; Bachman, Martin; Smollett, Katherine; Werner, Finn; Miska, Eric A.; Balasubramanian, Shankar (2015). "Formation and Abundance of 5-Hydroxymethylcytosine in RNA". ChemBioChem. 16 (5): 752–755. doi:10.1002/cbic.201500013. PMC 4471624. PMID 25676849.
- Delatte, B.; et al. (2016). "RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine". Science. 351 (6270): 282–285. doi:10.1126/science.aac5253. PMID 26816380.
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012; 13: 175. doi: 10.1186/gb-2012-13-10-175 Archived 2013-07-11 at the Wayback Machine.
- Garalde, DR; Snell, EA; Jachimowicz, D; Sipos, B; Lloyd, JH; Bruce, M; Pantic, N; et al. (2018). "Highly parallel direct RNA sequencing on an array of nanopores". Nature Methods. 15 (3): 201–206. doi:10.1038/nmeth.4577. PMID 29334379.