End-sequence profiling
End-sequence profiling (ESP) (sometimes "Paired-end mapping (PEM)") is a method based on sequence-tagged connectors developed to facilitate de novo genome sequencing to identify high-resolution copy number and structural aberrations such as inversions and translocations.
Briefly, the target genomic DNA is isolated and partially digested with restriction enzymes into large fragments. Following size-fractionation, the fragments are cloned into plasmids to construct artificial chromosomes such as bacterial artificial chromosomes (BAC) which are then sequenced and compared to the reference genome. The differences, including orientation and length variations between constructed chromosomes and the reference genome, will suggest copy number and structural aberration.
Artificial chromosome construction
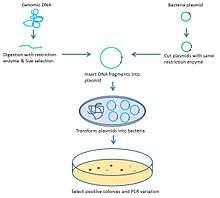
Before analyzing target genome structural aberration and copy number variation (CNV) with ESP, the target genome is usually amplified and conserved with artificial chromosome construction. The classic strategy to construct an artificial chromosome is bacterial artificial chromosome (BAC). Basically, the target chromosome is randomly digested and inserted into plasmids which are transformed and cloned in bacteria.[1] The size of fragments inserted is 150–350 kb.[2] Another commonly used artificial chromosome is fosmid. The difference between BAC and fosmids is the size of the DNA inserted. Fosmids can only hold 40 kb DNA fragments,[3] which allows a more accurate breakpoint determination.
Structural aberration detection
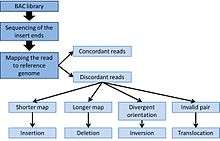
End sequence profiling (ESP) can be used to detect structural variations such as insertions, deletions, and chromosomal rearrangement. Compare to other methods that look at chromosomal abnormalities, ESP is particularly useful to identify copy neutral abnormalities such as inversions and translocations that would not be apparent when looking at copy number variation.[4][5] From the BAC library, both ends of the inserted fragments are sequenced using a sequencing platform. Detection of variations is then achieved by mapping the sequenced reads onto a reference genome.
Inversion and translocation
Inversions and translocations are relatively easy to detect by an invalid pair of sequenced-end. For instance, a translocation can be detected if the paired-ends are mapped onto different chromosomes on the reference genome.[4][5] Inversion can be detected by divergent orientation of the reads, where the insert will have two plus-end or two minus-end.
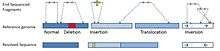
Insertion and deletion
In the case of an insertion or a deletion, mapping of the paired-end is consistent with the reference genome. But the read are disconcordant in apparent size. The apparent size is the distance of the BAC sequenced-ends mapped in the reference genome. If a BAC has an insert of length (l), a concordant mapping will show a fragment of size (l) in the reference genome. If the paired-ends are closer than distance (l), an insertion is suspected in the sampled DNA. A distance of (l< μ-3σ) can be used as a cut-off to detect an insertion, where μ is the mean length of the insert and σ is the standard deviation.[5][6] In case of a deletion, the paired-ends are mapped further away in the reference genome compared to the expected distance (l> μ-3σ).[6]
Copy number variation
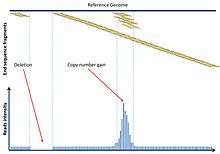
In some cases, discordant reads can also indicate a CNV for example in sequences repeats. For larger CNV, the density of the reads will vary accordingly to the copy number. An increase of copy numbers will be reflected by increasing mapping of the same region on the reference genome.
ESP history
ESP was first developed and published in 2003 by Dr. Collins and his colleagues in University of California, San Francisco. Their study revealed the chromosome rearrangements and CNV of MCF7 human cancer cells at a 150kb resolution, which is much more accurate compared to both CGH and spectral karyotyping at that time.[5] In 2007, Dr. Snyder and his group improved the ESP to 3kb resolution by sequencing both pairs of 3-kb DNA fragments without BAC construction. Their approach is able to identify deletions, inversions, insertions with an average breakpoint resolution of 644bp, which close to the resolution of polymerase chain reaction (PCR).[7]
ESP applications
Various bioinformatics tools can be used to analyze end-sequence profiling. Common ones include BreakDancer, PEMer, Variation Hunter, common LAW, GASV, and Spanner.[8] ESP can be used to map structural variation at high-resolution in disease tissue. This technique is mainly used on tumor samples from different cancer types. Accurate identification of copy neutral chromosomal abnormalities is particularly important as translocation can lead to fusion proteins, chimeric proteins, or misregulated proteins that can be seen in tumors. This technique can also be used in evolution studies by identifying large structural variation between different populations.[9] Similar methods are being developed for various applications. For example, a barcoded Illumina paired-end sequencing (BIPES) approach was used to assess microbial diversity by sequencing the 16S V6 tag.[10]
Advantages and limitations
Resolution of structural variation detection by ESP has been increased to the similar level as PCR, and can be further improved by selection of more evenly sized DNA fragments. ESP can be applied for either with or without constructed artificial chromosome. With BAC, precious samples can be immortalized and conserved, which is particularly important for small quantity of smalls which are planned for extensive analyses. Furthermore, BACs carrying rearranged DNA fragments can be directly transfected in vitro or in vivo to analyze the function of these arrangements. However, BAC construction is still expensive and labor-intensive. Researchers should be really careful to choose which strategy they need for particular project. Because ESP only looks at short paired-end sequences, it has the advantage of providing useful information genome-wide without the need for large-scale sequencing. Approximately 100-200 tumors can be sequenced at a resolution greater than 150kb when compared to sequencing an entire genome.
References
- O'Connor, M; Peifer, M; Bender, W (16 June 1989). "Construction of large DNA segments in Escherichia coli". Science. 244 (4910): 1307–12. Bibcode:1989Sci...244.1307O. doi:10.1126/science.2660262. PMID 2660262.
- Stone, NE; Fan, JB; Willour, V; Pennacchio, LA; Warrington, JA; Hu, A; de la Chapelle, A; Lehesjoki, AE; Cox, DR; Myers, RM (March 1996). "Construction of a 750-kb bacterial clone contig and restriction map in the region of human chromosome 21 containing the progressive myoclonus epilepsy gene". Genome Research. 6 (3): 218–25. doi:10.1101/gr.6.3.218. PMID 8963899.
- Tuzun, Eray; Sharp, Andrew J; Bailey, Jeffrey A; Kaul, Rajinder; Morrison, V Anne; Pertz, Lisa M; Haugen, Eric; Hayden, Hillary; Albertson, Donna; Pinkel, Daniel; Olson, Maynard V; Eichler, Evan E (15 May 2005). "Fine-scale structural variation of the human genome". Nature Genetics. 37 (7): 727–732. doi:10.1038/ng1562. PMID 15895083.
- Bashir, Ali; Volik, Stanislav; Collins, Colin; Bafna, Vineet; Raphael, Benjamin J.; Ouzounis, Christos A. (25 April 2008). "Evaluation of Paired-End Sequencing Strategies for Detection of Genome Rearrangements in Cancer". PLOS Computational Biology. 4 (4): e1000051. Bibcode:2008PLSCB...4E0051B. doi:10.1371/journal.pcbi.1000051. PMC 2278375. PMID 18404202.
- Volik, S.; Zhao, S.; Chin, K.; Brebner, J. H.; Herndon, D. R.; Tao, Q.; Kowbel, D.; Huang, G.; Lapuk, A.; Kuo, W.-L.; Magrane, G.; de Jong, P.; Gray, J. W.; Collins, C. (4 June 2003). "End-sequence profiling: Sequence-based analysis of aberrant genomes". Proceedings of the National Academy of Sciences. 100 (13): 7696–7701. Bibcode:2003PNAS..100.7696V. doi:10.1073/pnas.1232418100. PMC 164650. PMID 12788976.
- Yang, R; Chen, L; Newman, S; Gandhi, K; Doho, G; Moreno, CS; Vertino, PM; Bernal-Mizarchi, L; Lonial, S; Boise, LH; Rossi, M; Kowalski, J; Qin, ZS (2014). "Integrated analysis of whole-genome paired-end and mate-pair sequencing data for identifying genomic structural variations in multiple myeloma". Cancer Informatics. 13 (Suppl 2): 49–53. doi:10.4137/CIN.S13783. PMC 4179644. PMID 25288879.
- Korbel, JO; Urban, AE; Affourtit, JP; Godwin, B; Grubert, F; Simons, JF; Kim, PM; Palejev, D; Carriero, NJ; Du, L; Taillon, BE; Chen, Z; Tanzer, A; Saunders, AC; Chi, J; Yang, F; Carter, NP; Hurles, ME; Weissman, SM; Harkins, TT; Gerstein, MB; Egholm, M; Snyder, M (19 October 2007). "Paired-end mapping reveals extensive structural variation in the human genome". Science. 318 (5849): 420–6. Bibcode:2007Sci...318..420K. doi:10.1126/science.1149504. PMC 2674581. PMID 17901297.
- Zhao, Min; Wang, Qingguo; Wang, Quan; Jia, Peilin; Zhao, Zhongming (2013). "Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives". BMC Bioinformatics. 14 (Suppl 11): S1. doi:10.1186/1471-2105-14-S11-S1. PMC 3846878. PMID 24564169.
- Korbel, J. O.; Urban, A. E.; Affourtit, J. P.; Godwin, B.; Grubert, F.; Simons, J. F.; Kim, P. M.; Palejev, D.; Carriero, N. J.; Du, L.; Taillon, B. E.; Chen, Z.; Tanzer, A.; Saunders, A. C. E.; Chi, J.; Yang, F.; Carter, N. P.; Hurles, M. E.; Weissman, S. M.; Harkins, T. T.; Gerstein, M. B.; Egholm, M.; Snyder, M. (19 October 2007). "Paired-End Mapping Reveals Extensive Structural Variation in the Human Genome". Science. 318 (5849): 420–426. Bibcode:2007Sci...318..420K. doi:10.1126/science.1149504. PMC 2674581. PMID 17901297.
- Zhou, Hong-Wei; Li, Dong-Fang; Tam, Nora Fung-Yee; Jiang, Xiao-Tao; Zhang, Hai; Sheng, Hua-Fang; Qin, Jin; Liu, Xiao; Zou, Fei (21 October 2010). "BIPES, a cost-effective high-throughput method for assessing microbial diversity". The ISME Journal. 5 (4): 741–749. doi:10.1038/ismej.2010.160. PMC 3105743. PMID 20962877.