MECOM
MDS1 and EVI1 complex locus protein EVI1 (MECOM) also known as ecotropic virus integration site 1 protein homolog (EVI-1) or positive regulatory domain zinc finger protein 3 (PRDM3) is a protein that in humans is encoded by the MECOM gene. EVI1 was first identified as a common retroviral integration site in AKXD murine myeloid tumors. It has since been identified in a plethora of other organisms, and seems to play a relatively conserved developmental role in embryogenesis. EVI1 is a nuclear transcription factor involved in many signaling pathways for both coexpression and coactivation of cell cycle genes.

Gene structure
The EVI1 gene is located in the human genome on chromosome 3 (3q26.2). The gene spans 60 kilobases and encodes 16 exons, 10 of which are protein-coding. The first in-frame ATG start codon is in exon 3.[5]
mRNA
A large number of transcript variations exist, encoding different isoforms or chimeric proteins. Some of the most common ones are:
- EVI_1a, EVI_1b, EVI_1c, EVI_1d, and EVI_3L are all variants in the 5' untranslated region, and all except EVI_1a are specific to human cells.[6]
- −Rp9 variant is quite common in human and mouse cells, lacks 9 amino acids in the repression domain.[6]
- Δ324 found at low levels in human and mouse cells - an alternative splice variant encoding an 88kDa protein lacking zinc fingers 6 and 7 [6][7]
- Δ105 variant is unique to mice, and results in a protein truncated by 105 amino acids at the acidic C-terminus.[6]
- Fusion transcripts with upstream genes such as MDS1/EVI1 (ME), AML1/MDS1/EVI1 (AME), ETV6/MDS1/EVI1 have all been identified [6]
Protein
The MECOM is primarily found in the nucleus, either soluble or bound to DNA. The 145kDa isoform is the most-studied, encoding 1051 amino acids,[7] although there are many EVI1 fusion products detectable in cells expressing EVI1.
The MECOM protein contains 2 domains characterized by 7 zinc finger motifs followed by a proline-rich transcription repression domain, 3 more zinc finger motifs and an acidic C-terminus.[6]
Biological role
EVI1 is a proto-oncogene conserved across humans, mice, and rats, sharing 91% homology in nucleotide sequence and 94% homology in amino acid sequence between humans and mice.[7] It is a transcription factor localized to the nucleus and binds DNA through specific conserved sequences of GACAAGATA [8] with the potential to interact with both corepressors and coactivators.
- Embryogenesis
- The role of EVI1 in embryogenesis and development is not completely understood, but it has been shown that EVI1 deficiency in mice is an embryonic lethal mutation, characterized primarily by widespread hypocellularity and poor/disrupted development of the cardiovascular and neural system.[7] EVI1 is highly expressed in the murine embryo, found in the urinary system, lungs, and heart, but is only minutely detectable in most adult tissues,[7] indicating a likely role in tissue development. EVI1 and the fusion transcript MDS1-EVI1 are both expressed in the adult human kidney, lung, pancreas, brain, and ovaries.[7]
- Cell cycle and differentiation
- In vitro experiments using both human and mouse cell lines have shown that EVI1 prevents the terminal differentiation of bone marrow progenitor cells to granulocytes and erythroid cells, however it favors the differentiation of hematopoietic cell to megakaryocytes.[7] The chimeric gene of AML1-MDS1-EVI1 (AME) formed by the chromosomal translocation (3;21)(q26;q22) has also been shown in vitro to upregulate the cell cycle and block granulocytic differentiation of murine hematopoietic cells, as well as to delay the myeloid differentiation of bone marrow progenitors.[7]
Association with cancer
EVI1 has been described as a proto-oncogene since its first discovery in 1988.[9] Overexpression and aberrant expression of EVI1 has been associated with human acute myelogenous leukemia (AML), myelodysplastic syndrome (MDS) and chronic myelogenous leukemia (CML), and more recently has been shown as a poor prognostic indicator. Its function in these cells may be regulated by phosphorylation of serine196, in its N-terminal DNA binding domain.[10] All of these involve erratic cellular development and differentiation in the bone marrow leading to dramatic alterations in the normal population of blood cells. EVI1 has also been found to play a role in solid ovarian and colon tumors,[11] although it is not yet well characterized in this context. It has been hypothesized that it acts as a survival factor in tumor cell lines, preventing therapeutic-induced apoptosis and rendering the tumor cells more resistant to current treatments.[12]
Role in tumor suppressor signaling and prevention of apoptosis
TGF-β and cell cycle progression
EVI1 has been shown to be involved in the downstream signaling pathway of transforming growth factor beta (TGF-β). TGF-β, along with other TGF-β family ligands such as bone morphogenic protein (BMP) and activin are involved in regulating important cellular functions such as proliferation, differentiation, apoptosis, and matrix production.[13] These biological roles are not only important for cellular development, but also in understanding oncogenesis.
TGF-β signaling induces transcription of the cyclin-dependent kinase (CDK) inhibitors p15Ink4B or p21Cip1, which, as a consequence, act to halt the cell cycle and stop proliferation. This inhibition can lead to cellular differentiation or apoptosis, and therefore any resistance to TGF-β is thought to contribute in some way to human leukemogenesis.[14] As shown in the figure below, the downstream effectors of TGF-β are the Smad receptors (also known as receptor-activated Smads). Smad2 and Smad3 are phosphorylated in response to TGF-β ligand binding, and translocate into the nucleus of the cell, where they can then bind to DNA and other transcription factors.[13] Stable binding to promoters occurs through a conserved MH1 domain, and transcription activation occurs through an MH2 domain, and involves accompanying coactivators such as CBP/p300 and Sp1.[13]
The majority of literature discusses the interaction between EVI1 and Smad3, however there have been some experiments done showing that EVI1 interacts with all of the Smad proteins at varying levels, indicating a potential involvement in all of the pathways that include Smads as downstream effectors.[13] The translocation of phosphorylated Smad3 into the nucleus allows for direct interaction with EVI1, mediated by the first zinc finger domain on EVI1 and the MH2 domain on Smad3.[13][14] As the Smad3 MH2 domain is required for transcription activation, EVI1 binding effectively prevents transcription of the TGF-β induced anti-growth genes through structural blocking, and also leads to recruitment of other transcriptional repressors (see Epigenetics). By inhibiting an important checkpoint pathway for tumor suppression and growth control, overexpression or aberrant expression of EVI1 has characteristic oncogenic activity.
As an additional confirmation of the role of EVI1 expression on cell cycle progression, it has been shown that high EVI1 expression is correlated with the well-known tumor suppressor and cell cycle mediator Retinoblastoma, remaining in a hyperphosphorylated state, even in the presence of TGF-β.[15]
JNK and inhibition of apoptosis
c-Jun N-terminal kinase (JNK) is a MAP kinase activated by extracellular stress signals such as gamma-radiation, ultraviolet light, Fas ligand, tumor necrosis factor α (TNF-α), and interleukin-1.[16] Phosphorylation on two separate residues, Thr183 and Tyr185, cause JNK to become activated and translocate to the nucleus to phosphorylate and activate key transcription factors for the apoptotic response.[16]
Experiments co-expressing EVI1 and JNK have shown that levels of JNK-phosphorylated transcription factors (such as c-Jun) are drastically decreased in the presence of EVI1. Binding of EVI1 and JNK has been shown to occur through the first zinc finger motif on EVI1, and that this interaction does not block JNK phosphorylation and activation, but blocks JNK binding to substrate in the nucleus.[16] Subsequent in vitro assays showed that stress-induced cell death from a variety of stimuli is significantly inhibited by EVI1 and JNK binding.[16]
EVI1 does not bind other MAP kinases such as p38 or ERK.[16]
Oncogenesis and induced proliferation of HSCs
Among the many other observed defects, EVI1−/− mouse embryos have been shown to have defects in both the development and proliferation of hematopoietic stem cells (HSCs). It is presumed that this is due to direct interaction with the transcription factor GATA-2, which is crucial for HSC development.[17] It has subsequently been shown many times in vitro that EVI1 upregulation can induce proliferation and differentiation of HSCs and some other cell types such as rat fibroblasts.[6]
However, existing data is inconclusive regarding the absolute role of EVI1 in cell cycle progression. It appears to depend on the specific cell type, cell line and growth conditions being used as to whether EVI1 expression induces growth arrest or cell differentiation/proliferation, or whether it has any effect at all.[6] The data showing direct interaction of EVI1 with the promoters for a diverse array of genes supports the theory that this is a complex transcription factor associated with many different signalling pathways involved in development and growth.
Angiogenesis
Although the literature is limited on the subject, the well-documented effects on HSCs imply that there is a potential indirect effect of aberrant EVI1 expression on tumoral angiogenesis. HSCs secrete angiopoietin, and its receptor molecule Tie2 has been implicated in angiogenesis of tumors in both humans and mice.[18] Upregulation of Tie2 has been shown to occur under hypoxic conditions, and to increase angiogenesis when coinjected with tumor cells in mice.[18] Observations that EVI1−/− mutants have substantially downregulated Tie2 and Ang-I expression, therefore, hints at an interesting role of high EVI1 expression in tumor progression. This is likely, at least in part, a reason for the widespread hemorrhaging and minimal vascular development in EVI1 deleted embryos,[17] and has potential to indicate yet another reason for poor prognosis of EVI1 positive cancers.
Epigenetics
EVI1 has also been shown to directly interact with C-terminal-binding protein (CtBP, a known transcriptional repressor) through in vitro techniques such as yeast 2-hybrid screens and immunoprecipitation.[14] This interaction has been specifically shown to rely on amino acids 544-607 on the EVI1 protein, a stretch that contains two CtBP-binding consensus motifs.[15] This binding leads to recruitment of histone deacetylases (HDACs) as well as many other corepressor molecules leading to transcription repression via chromatin remodelling.[14]
EVI1 interaction with Smad3 followed by recruitment of corepressors can inhibit transcription and de-sensitize a cell to TGF-β signaling without ever displacing Smad3 from a gene's promoter.[13] The epigenetic modification is clearly enough to make the DNA inaccessible to the transcription machinery.
Although EVI1 has mainly been implicated as a transcription repressor, there is some data that has shown a possible dual role for this protein. Studies show that EVI1 also binds to known coactivators cAMP responsive element binding protein (CBP) and p300/CBP-associated factor (P/CAF).[13] These both have histone acetyltransferase activity, and lead to subsequent transcription activation. In addition, structural changes have been visualized within the nucleus of a cell, depending on the presence of corepressors or coactivators, leading researchers to believe that EVI1 has a unique response to each kind of molecule. In approximately 90% of cells, EVI1 is diffuse within the nucleus; however, when CBP and P/CAF are added, extensive nuclear speckle formation occurs.[19] The complete physiological repercussions of this complex role of EVI1 have yet to be elucidated, however, could provide insight into the wide variety of results that have been reported regarding the effect of EVI1 on in vitro cell proliferation.[6]
Interaction with corepressors and coactivators appears to occur in distinct domains,[19] and there are theories that EVI1 exists in a periodical, reversible acetylated state [7] within the cell. Contrasting theories indicate that the interplay between different EVI1 binding proteins acts to stabilize interactions with different transcription factors and DNA, leading to a response of EVI1 to a diverse set of stimuli.[13]
Chromosome instability
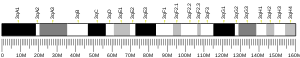
Since it was first identified in murine myeloid leukemia as a common site of retroviral integration into the chromosome, EVI1 and its surrounding DNA have been a site of many identified chromosomal translocations and abnormalities.[20] This can lead to aberrant expression of EVI1, and, as shown in the figure below, commonly involved chromosomal breakpoints have been mapped extensively. One major cause of EVI1 activation and consequent overexpression is a clinical condition called 3q21q26 syndrome from inv(3)(q21q26) or t(3;3)(q21;q26).[7] The result is the placement of a strong enhancing region for the housekeeping gene Ribophorin 1 (RPN1)[21] next to the EVI1 coding sequence, resulting in a dramatic increase of EVI1 levels in the cell.[7]
A summary of common chromosomal abnormalities involving EVI1 and its fusion genes can be found in a review by Nucifora et al..[22]
The most common circumstance involves chromosomal translocations in human AML or MDS, leading to constitutive expression of EVI1 and eventually to cancer.[22] Not only are these abnormalities in the 3q26 region associated with very poor patient prognosis they are also commonly accompanied by additional karyotypic changes such as chromosome 7 monosomy, deletion of the short arm of chromosome 7, or partial deletions of chromosome 5.[23] In addition, it has been shown that development of acute myelogenous leukemia is likely due to several sequential genetic changes, and that expression of EVI1 or its chimeric counterparts ME and AME alone is not enough to completely block myeloid differentiation.[24] BCR-Abl, a fusion gene caused by t(9;22)(q34;q11)is thought to have a cooperative effect with EVI1 during the progression of AML and CML.[24] Together, these two systems disrupt tyrosine kinase signaling and hematopoietic gene transcription.
Despite the extensively studied chromosomal abnormalities at the EVI1 locus, in anywhere from 10-50% of identified cases, EVI1 overexpression is detectable without any chromosomal abnormalities, indicating that there are other not-yet-understood systems, likely epigenetic, leading to EVI1 promoter activation.[6] In many of these cases, it is noted that a variety of 5' transcript variants are detectable at relatively high levels. Clinical studies have shown that these variants (EVI1_1a, EVI1_1b, EVI1_1d, EVI1_3L) as well as the MDS1-EVI1 fusion transcript are all associated with poor prognosis and increased likelihood of rapid remission in cases of de novo AML.[25]
Pharmacogenomics and cancer treatment
Very little research has been done in an attempt to therapeutically target EVI1 or any of its chimeric counterparts. However, since it has become an established fact that overexpression of EVI1 derivatives is a bad prognostic indicator, it is likely that the literature will begin to examine specific targeting within the next few years.
One very promising therapeutic agent for myelogenous leukemia and potentially other forms of cancer is arsenic trioxide (ATO). One study has been done showing that ATO treatment leads to specific degradation of the AML1/MDS1/EVI1 oncoprotein and induces both apoptosis and differentiation.[11] As an atypical use of traditional pharmacogenomics, this knowledge may lead to an increased ability to treat EVI1 positive leukemias that would normally have poor prognoses. If it is established that a clinical cancer case is EVI1 positive, altering the chemotherapeutic cocktail to include a specific EVI1 antagonist may aid to increase lifespan and prevent potential relapse. Arsenic is a fairly ancient human therapeutic agent,[11] however it has only recently returned to the forefront of cancer treatment. It has been observed that it not only induces apoptosis but can also inhibit the cell cycle, and has marked anti-angiogenesis effects.[26] As of 2006, Phase I and II clinical trials were being conducted to test this compound on a wide variety of cancer types, and currently (2008) a number of publications are showing positive outcomes in individual case studies, both pediatric and adult.
Hormones
The important and essential role of EVI1 in embryogenesis clearly indicates a close association with hormonal fluctuations in developing cells. However, to date, the presence of EVI1 in cancer has not been linked to aberrant production of any hormones or hormone receptors. It is likely that EVI1 is far enough downstream of hormonal signaling that once overproduced, it can function independently.
Future and current research
Effect on gene therapy
Areas where retroviral integration into the human genome is favored such as EVI1 have very important implications for the development of gene therapy. It was initially thought that delivery of genetic material through a non-replicating virus vector would pose no significant risk, as the likelihood of a random incorporation near a proto-oncogene was minimal. By 2008 it was realised that sites such as EVI1 are "highly over-represented" when it comes to vector insertions.[5]
Interactions
EVI1 has been shown to interact with:
- CREB binding protein,[19]
- CTBP1,[19][27]
- HDAC1,[19][28]
- Mothers against decapentaplegic homolog 3,[29] and
- PCAF[19] and
References
- GRCh38: Ensembl release 89: ENSG00000085276 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000027684 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Métais JY, Dunbar CE (Mar 2008). "The MDS1-EVI1 gene complex as a retrovirus integration site: impact on behavior of hematopoietic cells and implications for gene therapy". Molecular Therapy. 16 (3): 439–49. doi:10.1038/sj.mt.6300372. PMID 18227842.
- Wieser R (Jul 2007). "The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions". Gene. 396 (2): 346–57. doi:10.1016/j.gene.2007.04.012. PMID 17507183.
- Buonamici S, Chakraborty S, Senyuk V, Nucifora G (2003). "The role of EVI1 in normal and leukemic cells". Blood Cells, Molecules & Diseases. 31 (2): 206–12. doi:10.1016/S1079-9796(03)00159-1. PMID 12972028.
- Yatsula B, Lin S, Read AJ, Poholek A, Yates K, Yue D, Hui P, Perkins AS (Sep 2005). "Identification of binding sites of EVI1 in mammalian cells". The Journal of Biological Chemistry. 280 (35): 30712–22. doi:10.1074/jbc.M504293200. PMID 16006653.
- Morishita K, Parker DS, Mucenski ML, Jenkins NA, Copeland NG, Ihle JN (Sep 1988). "Retroviral activation of a novel gene encoding a zinc finger protein in IL-3-dependent myeloid leukemia cell lines". Cell. 54 (6): 831–40. doi:10.1016/S0092-8674(88)91175-0. PMID 2842066.
- White DJ, Unwin RD, Bindels E, Pierce A, Teng HY, Muter J, Greystoke B, Somerville TD, Griffiths J, Lovell S, Somervaille TC, Delwel R, Whetton AD, Meyer S (June 2013). "Phosphorylation of the leukemic oncoprotein EVI1 on serine 196 modulates DNA binding, transcriptional repression and transforming ability". PLOS ONE. 8 (6): e66510. Bibcode:2013PLoSO...866510W. doi:10.1371/journal.pone.0066510. PMC 3680417. PMID 23776681.
- Shackelford D, Kenific C, Blusztajn A, Waxman S, Ren R (Dec 2006). "Targeted degradation of the AML1/MDS1/EVI1 oncoprotein by arsenic trioxide". Cancer Research. 66 (23): 11360–9. doi:10.1158/0008-5472.CAN-06-1774. PMID 17145882.
- Liu Y, Chen L, Ko TC, Fields AP, Thompson EA (Jun 2006). "Evi1 is a survival factor which conveys resistance to both TGFbeta- and taxol-mediated cell death via PI3K/AKT". Oncogene. 25 (25): 3565–75. doi:10.1038/sj.onc.1209403. PMID 16462766.
- Alliston T, Ko TC, Cao Y, Liang YY, Feng XH, Chang C, Derynck R (Jun 2005). "Repression of bone morphogenetic protein and activin-inducible transcription by Evi-1". The Journal of Biological Chemistry. 280 (25): 24227–37. doi:10.1074/jbc.M414305200. PMID 15849193.
- Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H (May 2001). "The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling". Blood. 97 (9): 2815–22. doi:10.1182/blood.V97.9.2815. PMID 11313276.
- Hirai H, Izutsu K, Kurokawa M, Mitani K (Aug 2001). "Oncogenic mechanisms of Evi-1 protein". Cancer Chemotherapy and Pharmacology. 48 Suppl 1 (Suppl 1): S35-40. doi:10.1007/s002800100303. PMID 11587364. Archived from the original on 2013-02-12.
- Kurokawa M, Mitani K, Yamagata T, Takahashi T, Izutsu K, Ogawa S, Moriguchi T, Nishida E, Yazaki Y, Hirai H (Jun 2000). "The evi-1 oncoprotein inhibits c-Jun N-terminal kinase and prevents stress-induced cell death". The EMBO Journal. 19 (12): 2958–68. doi:10.1093/emboj/19.12.2958. PMC 203342. PMID 10856240.
- Yuasa H, Oike Y, Iwama A, Nishikata I, Sugiyama D, Perkins A, Mucenski ML, Suda T, Morishita K (Jun 2005). "Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression". The EMBO Journal. 24 (11): 1976–87. doi:10.1038/sj.emboj.7600679. PMC 1142611. PMID 15889140.
- De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE (Dec 2007). "Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications". Trends in Immunology. 28 (12): 519–24. doi:10.1016/j.it.2007.09.004. PMID 17981504.
- Chakraborty S, Senyuk V, Sitailo S, Chi Y, Nucifora G (Nov 2001). "Interaction of EVI1 with cAMP-responsive element-binding protein-binding protein (CBP) and p300/CBP-associated factor (P/CAF) results in reversible acetylation of EVI1 and in co-localization in nuclear speckles". The Journal of Biological Chemistry. 276 (48): 44936–43. doi:10.1074/jbc.M106733200. PMID 11568182.
- Morishita K, Parganas E, William CL, Whittaker MH, Drabkin H, Oval J, Taetle R, Valentine MB, Ihle JN (May 1992). "Activation of EVI1 gene expression in human acute myelogenous leukemias by translocations spanning 300-400 kilobases on chromosome band 3q26". Proceedings of the National Academy of Sciences of the United States of America. 89 (9): 3937–41. Bibcode:1992PNAS...89.3937M. doi:10.1073/pnas.89.9.3937. PMC 525606. PMID 1570317.
- "RPN1 ribophorin I [ Homo sapiens (human) ]". NCBI Resources. March 2013. Retrieved 21 March 2013.
- Nucifora G, Laricchia-Robbio L, Senyuk V (Mar 2006). "EVI1 and hematopoietic disorders: history and perspectives". Gene. 368: 1–11. doi:10.1016/j.gene.2005.09.020. PMID 16314052.
- Barjesteh van Waalwijk van Doorn-Khosrovani S, Erpelinck C, van Putten WL, Valk PJ, van der Poel-van de Luytgaarde S, Hack R, Slater R, Smit EM, Beverloo HB, Verhoef G, Verdonck LF, Ossenkoppele GJ, Sonneveld P, de Greef GE, Löwenberg B, Delwel R (Feb 2003). "High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients". Blood. 101 (3): 837–45. doi:10.1182/blood-2002-05-1459. PMID 12393383.
- Cuenco GM, Ren R (Dec 2001). "Cooperation of BCR-ABL and AML1/MDS1/EVI1 in blocking myeloid differentiation and rapid induction of an acute myelogenous leukemia". Oncogene. 20 (57): 8236–48. doi:10.1038/sj.onc.1205095. PMID 11781838.
- Haas K, Kundi M, Sperr WR, Esterbauer H, Ludwig WD, Ratei R, Koller E, Gruener H, Sauerland C, Fonatsch C, Valent P, Wieser R (Apr 2008). "Expression and prognostic significance of different mRNA 5'-end variants of the oncogene EVI1 in 266 patients with de novo AML: EVI1 and MDS1/EVI1 overexpression both predict short remission duration". Genes, Chromosomes & Cancer. 47 (4): 288–98. doi:10.1002/gcc.20532. PMID 18181178.
- Hu J, Fang J, Dong Y, Chen SJ, Chen Z (Feb 2005). "Arsenic in cancer therapy". Anti-Cancer Drugs. 16 (2): 119–27. doi:10.1097/00001813-200502000-00002. PMID 15655408.
- Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H (May 2001). "The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling". Blood. 97 (9): 2815–22. doi:10.1182/blood.v97.9.2815. PMID 11313276.
- Vinatzer U, Taplick J, Seiser C, Fonatsch C, Wieser R (Sep 2001). "The leukaemia-associated transcription factors EVI-1 and MDS1/EVI1 repress transcription and interact with histone deacetylase". British Journal of Haematology. 114 (3): 566–73. doi:10.1046/j.1365-2141.2001.02987.x. PMID 11552981.
- Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H (Jul 1998). "The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3". Nature. 394 (6688): 92–6. Bibcode:1998Natur.394...92K. doi:10.1038/27945. PMID 9665135.
Further reading
- Wieser R (Jul 2007). "The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions". Gene. 396 (2): 346–57. doi:10.1016/j.gene.2007.04.012. PMID 17507183.
- Morishita K, Parganas E, Douglass EC, Ihle JN (Jul 1990). "Unique expression of the human Evi-1 gene in an endometrial carcinoma cell line: sequence of cDNAs and structure of alternatively spliced transcripts". Oncogene. 5 (7): 963–71. PMID 2115646.
- Mitani K, Ogawa S, Tanaka T, Miyoshi H, Kurokawa M, Mano H, Yazaki Y, Ohki M, Hirai H (Feb 1994). "Generation of the AML1-EVI-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia". The EMBO Journal. 13 (3): 504–10. doi:10.1002/j.1460-2075.1994.tb06288.x. PMC 394839. PMID 8313895.
- Perkins AS, Kim JH (Jan 1996). "Zinc fingers 1-7 of EVI1 fail to bind to the GATA motif by itself but require the core site GACAAGATA for binding". The Journal of Biological Chemistry. 271 (2): 1104–10. doi:10.1074/jbc.271.2.1104. PMID 8557637.
- Fears S, Mathieu C, Zeleznik-Le N, Huang S, Rowley JD, Nucifora G (Feb 1996). "Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family". Proceedings of the National Academy of Sciences of the United States of America. 93 (4): 1642–7. Bibcode:1996PNAS...93.1642F. doi:10.1073/pnas.93.4.1642. PMC 39995. PMID 8643684.
- Ogawa S, Kurokawa M, Tanaka T, Mitani K, Inazawa J, Hangaishi A, Tanaka K, Matsuo Y, Minowada J, Tsubota T, Yazaki Y, Hirai H (Jul 1996). "Structurally altered Evi-1 protein generated in the 3q21q26 syndrome". Oncogene. 13 (1): 183–91. PMID 8700545.
- Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H (Jul 1998). "The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3". Nature. 394 (6688): 92–6. Bibcode:1998Natur.394...92K. doi:10.1038/27945. PMID 9665135.
- Turner J, Crossley M (Sep 1998). "Cloning and characterization of mCtBP2, a co-repressor that associates with basic Krüppel-like factor and other mammalian transcriptional regulators". The EMBO Journal. 17 (17): 5129–40. doi:10.1093/emboj/17.17.5129. PMC 1170841. PMID 9724649.
- Kurokawa M, Mitani K, Yamagata T, Takahashi T, Izutsu K, Ogawa S, Moriguchi T, Nishida E, Yazaki Y, Hirai H (Jun 2000). "The evi-1 oncoprotein inhibits c-Jun N-terminal kinase and prevents stress-induced cell death". The EMBO Journal. 19 (12): 2958–68. doi:10.1093/emboj/19.12.2958. PMC 203342. PMID 10856240.
- Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H (May 2001). "The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling". Blood. 97 (9): 2815–22. doi:10.1182/blood.V97.9.2815. PMID 11313276.
- Palmer S, Brouillet JP, Kilbey A, Fulton R, Walker M, Crossley M, Bartholomew C (Jul 2001). "Evi-1 transforming and repressor activities are mediated by CtBP co-repressor proteins". The Journal of Biological Chemistry. 276 (28): 25834–40. doi:10.1074/jbc.M102343200. PMID 11328817.
- Chakraborty S, Senyuk V, Sitailo S, Chi Y, Nucifora G (Nov 2001). "Interaction of EVI1 with cAMP-responsive element-binding protein-binding protein (CBP) and p300/CBP-associated factor (P/CAF) results in reversible acetylation of EVI1 and in co-localization in nuclear speckles". The Journal of Biological Chemistry. 276 (48): 44936–43. doi:10.1074/jbc.M106733200. PMID 11568182.
- Shimizu S, Nagasawa T, Katoh O, Komatsu N, Yokota J, Morishita K (Apr 2002). "EVI1 is expressed in megakaryocyte cell lineage and enforced expression of EVI1 in UT-7/GM cells induces megakaryocyte differentiation". Biochemical and Biophysical Research Communications. 292 (3): 609–16. doi:10.1006/bbrc.2002.6693. PMID 11922610.
- Barjesteh van Waalwijk van Doorn-Khosrovani S, Erpelinck C, van Putten WL, Valk PJ, van der Poel-van de Luytgaarde S, Hack R, Slater R, Smit EM, Beverloo HB, Verhoef G, Verdonck LF, Ossenkoppele GJ, Sonneveld P, de Greef GE, Löwenberg B, Delwel R (Feb 2003). "High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients". Blood. 101 (3): 837–45. doi:10.1182/blood-2002-05-1459. PMID 12393383.
- Vinatzer U, Mannhalter C, Mitterbauer M, Gruener H, Greinix H, Schmidt HH, Fonatsch C, Wieser R (Jan 2003). "Quantitative comparison of the expression of EVI1 and its presumptive antagonist, MDS1/EVI1, in patients with myeloid leukemia". Genes, Chromosomes & Cancer. 36 (1): 80–9. doi:10.1002/gcc.10144. PMID 12461752.
- Chi Y, Senyuk V, Chakraborty S, Nucifora G (Dec 2003). "EVI1 promotes cell proliferation by interacting with BRG1 and blocking the repression of BRG1 on E2F1 activity". The Journal of Biological Chemistry. 278 (50): 49806–11. doi:10.1074/jbc.M309645200. PMID 14555651.
- Alliston T, Ko TC, Cao Y, Liang YY, Feng XH, Chang C, Derynck R (Jun 2005). "Repression of bone morphogenetic protein and activin-inducible transcription by Evi-1". The Journal of Biological Chemistry. 280 (25): 24227–37. doi:10.1074/jbc.M414305200. PMID 15849193.
- Nitta E, Izutsu K, Yamaguchi Y, Imai Y, Ogawa S, Chiba S, Kurokawa M, Hirai H (Sep 2005). "Oligomerization of Evi-1 regulated by the PR domain contributes to recruitment of corepressor CtBP". Oncogene. 24 (40): 6165–73. doi:10.1038/sj.onc.1208754. PMID 15897867.
- Maki K, Yamagata T, Asai T, Yamazaki I, Oda H, Hirai H, Mitani K (Sep 2005). "Dysplastic definitive hematopoiesis in AML1/EVI1 knock-in embryos". Blood. 106 (6): 2147–55. doi:10.1182/blood-2004-11-4330. PMID 15914564.