CpG island hypermethylation
CpG island hypermethylation is an epigenetic control aberration that is important for gene inactivation in cancer cells. Hypermethylation of CpG islands has been described in almost every type of tumor. Many important cellular pathways, such as DNA repair (hMLH1, for example), cell cycle (p14ARF), apoptosis (DAPK), cell adherence (CDH1, CDH13), are inactivated by this epigenetic lesion.[1] Hypermethylation is linked to methyl-binding proteins, DNA methyltransferases and histone deacetylase, but the degree to which this process selectively silences tumor suppressor genes remains a vibrant field of study. The list for hypermethylated genes is growing and functional and genetic studies are being performed to determine which hypermethylation events are relevant for tumorigenesis. Basic as well as translational research will be needed to understand the mechanisms and roles of CpG island hypermethylation in cancer.
History
The first discovery of methylation in a CpG island of a tumor suppressor gene in humans was that of the Retinoblastoma (Rb) gene in 1989.[2] This was just a few years after the first oncogene mutation was discovered in a human primary tumor. The discovery of the methylation-associated inactivation of the Von Hippel-Lindau (VHL) gene revived the idea of the hypermethylation of the CpG island promoter being a mechanism to inactivate genes in cancer.[3] Cancer epigenetic silencing in its current state was born in the labs of Baylin and Jones,[3] where it was proven that CpG island hypermethylation was a common inactivation mechanism of the tumor suppressor gene p16INK4a. The introduction of methylation-specific PCR and sodium bisulfite modification added tools to the belt of cancer epigenetics research,[3][4] and the list of candidate genes with aberrant methylation of their CpG islands has been growing since.[5] Initially, the presence of alterations in the profile of DNA methylation in cancer was seen as a global hypomethylation of the genome that would lead to massive overexpression of oncogenes with a normally hypermethylated CpG island.[6] Lately, this is considered as an incomplete scenario, despite the idea of the genome of the cancer cell undergoing a reduction of its 5-methylcytosine content when compared to its parent normal cell being correct.[5] In normal tissues, the vast majority of CpG islands are completely unmethylated with some exceptions.[1] The association of transcriptional silencing of tumor suppressor genes with hypermethylation is the foundation upon which this subset of cancer epigenetics stands.

Structure
In a normal cell, the CpG island is hypomethylated,[7] and the rest of the genome is methylated. It is evident that the hypomethylation of the CpG island in normal cells provides no additional steric hindrance to future binding. The majority of CpG pairs in mammals are chemically modified by the covalent attachment of a methyl group to the C5 position of the cytosine ring.[8] This modification is distributed throughout the genome and represses transcription. A CpG island is a Cytosine and Guanine linked by a phosphate in a repeated sequence. These are genetic hotspots as they are sites for active methylation. The expression of a gene is tissue specific, which leads to variation in tissue function. Methylation of a gene prevents expression of a gene in a particular way.

The reason for methylation to be almost exclusive to CpG dinucleotides is the symmetry of the dinucleotide. This allows for preservation during cell division and is a hallmark for epigenetic modifications.

Role in cancer
In order to understand the role of CpG island hypermethylation in cancer, it is useful to consider a particular tumor type, called CpG island methylator prototype, or CIMP. Higher levels of CpG island hypermethylation are found in CIMP. The frequent occurrence of hypermethylation was first described in colorectal cancer and later for glioma. More recently, it has been studied for neuroblastomas. The frequent hypermethylation of CpG islands in these tumors as well as their irregularity are ways to determine that hypermethylated CpGs differ by tumor type. This would imply that colorectal cancer will not necessarily have the same set of hypermethylated CpG islands as in a glioma. This clinical distinctness of tumors can be interpreted by doctors. To zoom in further, let us focus on colorectal CIMP, as this was one of the first of its kind to be described. Patients in this category of cancer tend to be older and female and have a defective MLH1 function. The tumors are usually in the ascending colon. They also have a good prognostic outcome, so if a patient were diagnosed with this variety of CIMP, they are likely to have a good outcome. These clinically distinct phenotypes of CIMP also suggest that there is potential for epigenetic therapy. CpG islands that are hypermethylated can play three roles in cancer: in diagnosis, prognosis and in monitoring. In diagnosis, one can identify the tumor type and tumor subtype, as well as its primary tumor when that is unknown. Hypermethylation increases with tumorigenicity, which is an indication of the prognosis of cancer. For example, high methylation is a marker for poor prognosis in lung cancer. CpG island hypermethylation shows much promise for molecular monitoring of patients with cancer. It is also a potential target for therapeutic use.
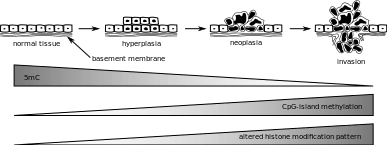
Aberrations in epigenetic control that are seen in cancer pertain to DNA methylation, which can be either locus-specific DNA hypermethylation or genome-wide DNA hypomethylation. Under locus-specific DNA hypermethylation comes CpG island hypermethylation. DNA methylation acts as an alternative to genetic mutation. According to the Knudson hypothesis, cancer is a result of multiple hits to DNA, and DNA methylation can be one such hit. Epigenetic mutations such as DNA methylation are mitotically heritable, but also reversible, unlike gene mutations. The identity of hypermethylated CpG islands varies by the type of tumor. Some single gene examples include MLH1 in colorectal cancer and BRCA1 in breast cancer. Hypermethylated CpG islands also act as biomarkers, as they can help distinguish cancer from normal cells in the same sample. They can also help identify certain specific features of cancer and their detection is more sensitive. CpG island hypermethylation has shown promise for the molecular monitoring of patients with cancer and is a potential target for therapeutic use.
References
- Esteller M (12 August 2002). "CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future". Oncogene. 21 (35): 5427–40. doi:10.1038/sj.onc.1205600. PMID 12154405. ProQuest 227311892.
- Greger V, Passarge E, Höpping W, Messmer E, Horsthemke B (1 September 1989). "Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma". Human Genetics. 83 (2): 155–8. doi:10.1007/BF00286709. PMID 2550354.
- Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM (11 October 1994). "Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma". Proceedings of the National Academy of Sciences. 91 (21): 9700–4. doi:10.1073/pnas.91.21.9700. PMC 44884. PMID 7937876.
- Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, Gusterson BA, Cooper CS (1 August 1994). "Identification of novel genes, SYT and SSX, involved in the t (X; 18)(p11. 2; q11. 2) translocation found in human synovial sarcoma". Nature Genetics. 7 (4): 502–8. doi:10.1038/ng0894-502. PMID 7951320.
- Esteller M, Corn PG, Baylin SB, Herman JG (15 April 2001). "A gene hypermethylation profile of human cancer". Cancer Research. 61 (8): 3225–9. PMID 11309270.
- Feinberg AP, Vogelstein B (1 July 1983). "A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity". Analytical Biochemistry. 132 (1): 6–13. doi:10.1016/0003-2697(83)90418-9. PMID 6312838.
- University of Melbourne, Coursera
- Illingworth RS, Bird AP (5 June 2009). "CpG islands–'a rough guide'". FEBS Letters. 583 (11): 1713–20. doi:10.1016/j.febslet.2009.04.012. PMID 19376112.