CpG site
The CpG sites or CG sites are regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide in the linear sequence of bases along its 5' → 3' direction. CpG sites occur with high frequency in genomic regions called CpG islands (or CG islands). Cytosines in CpG dinucleotides can be methylated to form 5-methylcytosines. Enzymes that add a methyl group are called DNA methyltransferases. In mammals, 70% to 80% of CpG cytosines are methylated.[1] Methylating the cytosine within a gene can change its expression, a mechanism that is part of a larger field of science studying gene regulation that is called epigenetics.
| CpG sites | GpC sites |
|---|---|
 |  |
| Distribution of CpG sites (left: in red) and GpC sites (right: in green) in the human APRT gene. CpG are more abundant in the upstream region of the gene where they form a CpG island, whereas GpC are more evenly distributed. The 5 exons of the APRT gene are indicated (blue), and the start (ATG) and stop (TGA) codons are emphasized (bold blue). | |

CpG characteristics
Definition
CpG is shorthand for 5'—C—phosphate—G—3' , that is, cytosine and guanine separated by only one phosphate group; phosphate links any two nucleosides together in DNA. The CpG notation is used to distinguish this single-stranded linear sequence from the CG base-pairing of cytosine and guanine for double-stranded sequences. The CpG notation is therefore to be interpreted as the cytosine being 5 prime to the guanine base. CpG should not be confused with GpC, the latter meaning that a guanine is followed by a cytosine in the 5' → 3' direction of a single-stranded sequence.
Under-representation
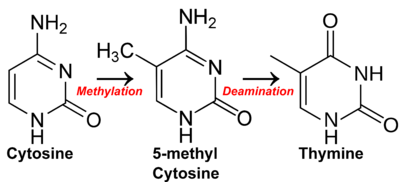
CpG dinucleotides have long been observed to occur with a much lower frequency in the sequence of vertebrate genomes than would be expected due to random chance. For example, in the human genome, which has a 42% GC content,[2] a pair of nucleotides consisting of cytosine followed by guanine would be expected to occur 0.21 * 0.21 = 4.41% of the time. The frequency of CpG dinucleotides in human genomes is 0.98%, less than one-quarter of the expected frequency.[3] This underrepresentation is a consequence of the high mutation rate of methylated CpG sites: the spontaneously occurring deamination of a methylated cytosine results in a thymine, and the resulting G:T mismatched bases are often improperly resolved to A:T; whereas the deamination of cytosine results in a uracil, which as a foreign base is quickly replaced by a cytosine by the base excision repair mechanism. The C to T transition rate at methylated CpG sites is ~10 fold higher than at unmethylated sites.[4][5][6][7]
Genomic distribution
CpG dinucleotides frequently occur in CpG islands (see definition of CpG islands, below). There are 28,890 CpG islands in the human genome, (50,267 if one includes CpG islands in repeat sequences).[8] This is in agreement with the 28,519 CpG islands found by Venter et al.[9] since the Venter et al. genome sequence did not include the interiors of highly similar repetitive elements and the extremely dense repeat regions near the centromeres.[10] Since CpG islands contain multiple CpG dinucleotide sequences, there appear to be more than 20 million CpG dinucleotides in the human genome.
CpG islands
CpG islands (or CG islands) are regions with a high frequency of CpG sites. Though objective definitions for CpG islands are limited, the usual formal definition is a region with at least 200 bp, a GC percentage greater than 50%, and an observed-to-expected CpG ratio greater than 60%. The "observed-to-expected CpG ratio" can be derived where the observed is calculated as:

and the expected as:

or

Many genes in mammalian genomes have CpG islands associated with the start of the gene[13] (promoter regions). Because of this, the presence of a CpG island is used to help in the prediction and annotation of genes.
In mammalian genomes, CpG islands are typically 300-3,000 base pairs in length, and have been found in or near approximately 40% of promoters of mammalian genes.[14] About 70% of human promoters have a high CpG content. Given the frequency of GC two-nucleotide sequences, the number of CpG dinucleotides is much lower than would be expected.[12]
A 2002 study revised the rules of CpG island prediction to exclude other GC-rich genomic sequences such as Alu repeats. Based on an extensive search on the complete sequences of human chromosomes 21 and 22, DNA regions greater than 500 bp were found more likely to be the "true" CpG islands associated with the 5' regions of genes if they had a GC content greater than 55%, and an observed-to-expected CpG ratio of 65%.[15]
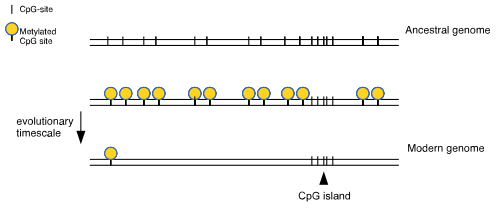
CpG islands are characterized by CpG dinucleotide content of at least 60% of that which would be statistically expected (~4–6%), whereas the rest of the genome has much lower CpG frequency (~1%), a phenomenon called CG suppression. Unlike CpG sites in the coding region of a gene, in most instances the CpG sites in the CpG islands of promoters are unmethylated if the genes are expressed. This observation led to the speculation that methylation of CpG sites in the promoter of a gene may inhibit gene expression. Methylation, along with histone modification, is central to imprinting.[16] Most of the methylation differences between tissues, or between normal and cancer samples, occur a short distance from the CpG islands (at "CpG island shores") rather than in the islands themselves.[17]
CpG islands typically occur at or near the transcription start site of genes, particularly housekeeping genes, in vertebrates.[12] A C (cytosine) base followed immediately by a G (guanine) base (a CpG) is rare in vertebrate DNA because the cytosines in such an arrangement tend to be methylated. This methylation helps distinguish the newly synthesized DNA strand from the parent strand, which aids in the final stages of DNA proofreading after duplication. However, over time methylated cytosines tend to turn into thymines because of spontaneous deamination. There is a special enzyme in humans (Thymine-DNA glycosylase, or TDG) that specifically replaces T's from T/G mismatches. However, due to the rarity of CpGs, it is theorised to be insufficiently effective in preventing a possibly rapid mutation of the dinucleotides. The existence of CpG islands is usually explained by the existence of selective forces for relatively high CpG content, or low levels of methylation in that genomic area, perhaps having to do with the regulation of gene expression. A 2011 study showed that most CpG islands are a result of non-selective forces.[18]
Methylation, silencing, cancer, and aging
CpG islands in promoters
In humans, about 70% of promoters located near the transcription start site of a gene (proximal promoters) contain a CpG island.[19][20]
Distal promoter elements also frequently contain CpG islands. An example is the DNA repair gene ERCC1, where the CpG island-containing element is located about 5,400 nucleotides upstream of the transcription start site of the ERCC1 gene.[21] CpG islands also occur frequently in promoters for functional noncoding RNAs such as microRNAs.[22]
Methylation of CpG islands stably silences genes
In humans, DNA methylation occurs at the 5 position of the pyrimidine ring of the cytosine residues within CpG sites to form 5-methylcytosines. The presence of multiple methylated CpG sites in CpG islands of promoters causes stable silencing of genes.[23] Silencing of a gene may be initiated by other mechanisms, but this is often followed by methylation of CpG sites in the promoter CpG island to cause the stable silencing of the gene.[23]
Promoter CpG hyper/hypo-methylation in cancer
In cancers, loss of expression of genes occurs about 10 times more frequently by hypermethylation of promoter CpG islands than by mutations. For example, in a colorectal cancer there are usually about 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations.[24] In contrast, in one study of colon tumors compared to adjacent normal-appearing colonic mucosa, 1,734 CpG islands were heavily methylated in tumors whereas these CpG islands were not methylated in the adjacent mucosa.[25] Half of the CpG islands were in promoters of annotated protein coding genes,[25] suggesting that about 867 genes in a colon tumor have lost expression due to CpG island methylation. A separate study found an average of 1,549 differentially methylated regions (hypermethylated or hypomethylated) in the genomes of six colon cancers (compared to adjacent mucosa), of which 629 were in known promoter regions of genes.[26] A third study found more than 2,000 genes differentially methylated between colon cancers and adjacent mucosa. Using gene set enrichment analysis, 569 out of 938 gene sets were hypermethylated and 369 were hypomethylated in cancers.[27] Hypomethylation of CpG islands in promoters results in overexpression of the genes or gene sets affected.
One 2012 study[28] listed 147 specific genes with colon cancer-associated hypermethylated promoters, along with the frequency with which these hypermethylations were found in colon cancers. At least 10 of those genes had hypermethylated promoters in nearly 100% of colon cancers. They also indicated 11 microRNAs whose promoters were hypermethylated in colon cancers at frequencies between 50% and 100% of cancers. MicroRNAs (miRNAs) are small endogenous RNAs that pair with sequences in messenger RNAs to direct post-transcriptional repression. On average, each microRNA represses several hundred target genes.[29] Thus microRNAs with hypermethylated promoters may be allowing over-expression of hundreds to thousands of genes in a cancer.
The information above shows that, in cancers, promoter CpG hyper/hypo-methylation of genes and of microRNAs causes loss of expression (or sometimes increased expression) of far more genes than does mutation.
DNA repair genes with hyper/hypo-methylated promoters in cancers
DNA repair genes are frequently repressed in cancers due to hypermethylation of CpG islands within their promoters. In head and neck squamous cell carcinomas at least 15 DNA repair genes have frequently hypermethylated promoters; these genes are XRCC1, MLH3, PMS1, RAD51B, XRCC3, RAD54B, BRCA1, SHFM1, GEN1, FANCE, FAAP20, SPRTN, SETMAR, HUS1, and PER1.[30] About seventeen types of cancer are frequently deficient in one or more DNA repair genes due to hypermethylation of their promoters.[31] As an example, promoter hypermethylation of the DNA repair gene MGMT occurs in 93% of bladder cancers, 88% of stomach cancers, 74% of thyroid cancers, 40%-90% of colorectal cancers and 50% of brain cancers. Promoter hypermethylation of LIG4 occurs in 82% of colorectal cancers. Promoter hypermethylation of NEIL1 occurs in 62% of head and neck cancers and in 42% of non-small-cell lung cancers. Promoter hypermethylation of ATM occurs in 47% of non-small-cell lung cancers. Promoter hypermethylation of MLH1 occurs in 48% of non-small-cell lung cancer squamous cell carcinomas. Promoter hypermethylation of FANCB occurs in 46% of head and neck cancers.
On the other hand, the promoters of two genes, PARP1 and FEN1, were hypomethylated and these genes were over-expressed in numerous cancers. PARP1 and FEN1 are essential genes in the error-prone and mutagenic DNA repair pathway microhomology-mediated end joining. If this pathway is over-expressed the excess mutations it causes can lead to cancer. PARP1 is over-expressed in tyrosine kinase-activated leukemias,[32] in neuroblastoma,[33] in testicular and other germ cell tumors,[34] and in Ewing's sarcoma,[35] FEN1 is over-expressed in the majority of cancers of the breast,[36] prostate,[37] stomach,[38][39] neuroblastomas,[40] pancreatic,[41] and lung.[42]
DNA damage appears to be the primary underlying cause of cancer.[43][44] If accurate DNA repair is deficient, DNA damages tend to accumulate. Such excess DNA damage can increase mutational errors during DNA replication due to error-prone translesion synthesis. Excess DNA damage can also increase epigenetic alterations due to errors during DNA repair.[45][46] Such mutations and epigenetic alterations can give rise to cancer (see malignant neoplasms). Thus, CpG island hyper/hypo-methylation in the promoters of DNA repair genes are likely central to progression to cancer.
Methylation of CpG sites with age
Since age has a strong effect on DNA methylation levels on tens of thousands of CpG sites, one can define a highly accurate biological clock (referred to as epigenetic clock or DNA methylation age) in humans and chimpanzees.[47]
Unmethylated sites
Unmethylated CpG dinucleotide sites can be detected by Toll-like receptor 9[48] (TLR 9) on plasmacytoid dendritic cells, monocytes, natural killer (NK) cells, and B cells in humans. This is used to detect intracellular viral infection.
Role of CpG sites in memory
In mammals, DNA methyltransferases (which add methyl groups to DNA bases) exhibit a sequence preference for cytosines within CpG sites.[49] In the mouse brain, 4.2% of all cytosines are methylated, primarily in the context of CpG sites, forming 5mCpG.[50] Most hypermethylated 5mCpG sites increase the repression of associated genes.[50]
As reviewed by Duke et al., neuron DNA methylation (repressing expression of particular genes) is altered by neuronal activity. Neuron DNA methylation is required for synaptic plasticity; is modified by experiences; and active DNA methylation and demethylation is required for memory formation and maintenance.[51]
In 2016 Halder et al.[52] using mice, and in 2017 Duke et al.[51] using rats, subjected the rodents to contextual fear conditioning, causing an especially strong long-term memory to form. At 24 hours after the conditioning, in the hippocampus brain region of rats, the expression of 1,048 genes was down-regulated (usually associated with 5mCpG in gene promoters) and the expression of 564 genes was up-regulated (often associated with hypomethylation of CpG sites in gene promoters). At 24 hours after training, 9.2% of the genes in the rat genome of hippocampus neurons were differentially methylated. However while the hippocampus is essential for learning new information it does not store information itself. In the mouse experiments of Halder, 1,206 differentially methylated genes were seen in the hippocampus one hour after contextual fear conditioning but these altered methylations were reversed and not seen after four weeks. In contrast with the absence of long-term CpG methylation changes in the hippocampus, substantial differential CpG methylation could be detected in cortical neurons during memory maintenance. There were 1,223 differentially methylated genes in the anterior cingulate cortex of mice four weeks after contextual fear conditioning.
Demethylation at CpG sites requires ROS activity

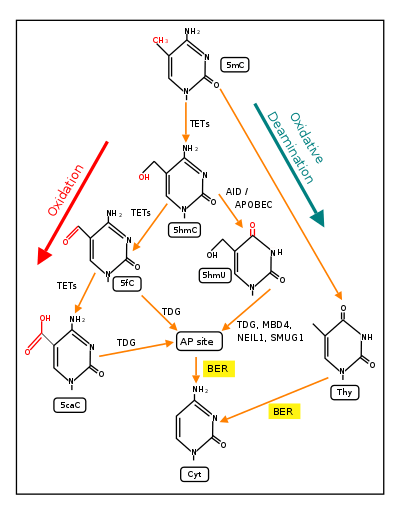
Two reviews[55][56] summarize the large body of evidence for the critical and essential role of ROS in memory formation. The DNA demethylation of thousands of CpG sites during memory formation depends on initiation by ROS. In 2016, Zhou et al.,[53] showed that ROS have a central role in DNA demethylation.
TET1 is a key enzyme involved in demethylating 5mCpG. However, TET1 is only able to act on 5mCpG if an ROS has first acted on the guanine to form 8-hydroxy-2'-deoxyguanosine (8-OHdG), resulting in a 5mCp-8-OHdG dinucleotide (see first figure in this section).[53] After formation of 5mCp-8-OHdG, the base excision repair enzyme OGG1 binds to the 8-OHdG lesion without immediate excision. Adherence of OGG1 to the 5mCp-8-OHdG site recruits TET1, allowing TET1 to oxidize the 5mC adjacent to 8-OHdG, as shown in the first figure in this section. This initiates the demethylation pathway shown in the second figure in this section.
Altered protein expression in neurons, controlled by ROS-dependent demethylation of CpG sites in gene promoters within neuron DNA, is central to memory formation.[57]
See also
- TLR9, detector of unmethylated CpG sites
- DNA methylation age
References
- Jabbari K, Bernardi G (May 2004). "Cytosine methylation and CpG, TpG (CpA) and TpA frequencies". Gene. 333: 143–9. doi:10.1016/j.gene.2004.02.043. PMID 15177689.
- Lander, Eric S.; Linton, Lauren M.; Birren, Bruce; Nusbaum, Chad; Zody, Michael C.; Baldwin, Jennifer; Devon, Keri; Dewar, Ken; Doyle, Michael (15 February 2001). "Initial sequencing and analysis of the human genome". Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. ISSN 1476-4687. PMID 11237011.
- stevens M, Cheng J, Li D, Xi M, Hong C, Maire C, Ligon K, Hirst M, Marra M, Costello J, Wang T (2013). "Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods". Genome Research. 23 (9): 1541–1553. doi:10.1101/gr.152231.112. PMC 3759729. PMID 23804401.
- Hwang DG, Green P (2004). "Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution". Proc Natl Acad Sci U S A. 101 (39): 13994–4001. Bibcode:2004PNAS..10113994H. doi:10.1073/pnas.0404142101. PMC 521089. PMID 15292512.
- Walsh CP, Xu GL (2006). "Cytosine methylation and DNA repair". Curr Top Microbiol Immunol. Current Topics in Microbiology and Immunology. 301: 283–315. doi:10.1007/3-540-31390-7_11. ISBN 3-540-29114-8. PMID 16570853.
- Arnheim N, Calabrese P (2009). "Understanding what determines the frequency and pattern of human germline mutations". Nat Rev Genet. 10 (7): 478–488. doi:10.1038/nrg2529. PMC 2744436. PMID 19488047.
- Ségurel L, Wyman MJ, Przeworski M (2014). "Understanding what determines the frequency and pattern of human germline mutations". Annu Rev Genom Hum Genet. 15: 47–70. doi:10.1146/annurev-genom-031714-125740. PMID 25000986.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Szustakowki J (February 2001). "Initial sequencing and analysis of the human genome". Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. PMID 11237011.
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X (February 2001). "The sequence of the human genome". Science. 291 (5507): 1304–51. Bibcode:2001Sci...291.1304V. doi:10.1126/science.1058040. PMID 11181995.
- Myers EW, Sutton GG, Smith HO, Adams MD, Venter JC (April 2002). "On the sequencing and assembly of the human genome". Proc. Natl. Acad. Sci. U.S.A. 99 (7): 4145–6. Bibcode:2002PNAS...99.4145M. doi:10.1073/pnas.092136699. PMC 123615. PMID 11904395.
- Gardiner-Garden M, Frommer M (1987). "CpG islands in vertebrate genomes". Journal of Molecular Biology. 196 (2): 261–282. doi:10.1016/0022-2836(87)90689-9. PMID 3656447.
- Saxonov S, Berg P, Brutlag DL (2006). "A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters". Proc Natl Acad Sci USA. 103 (5): 1412–1417. Bibcode:2006PNAS..103.1412S. doi:10.1073/pnas.0510310103. PMC 1345710. PMID 16432200.
- Hartl DL, Jones EW (2005). Genetics: Analysis of Genes and Genomes (6th ed.). Missisauga: Jones & Bartlett, Canada. p. 477. ISBN 978-0-7637-1511-3.
- Fatemi M, Pao MM, Jeong S, Gal-Yam EN, Egger G, Weisenberger DJ, Jones PA (2005). "Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level". Nucleic Acids Res. 33 (20): e176. doi:10.1093/nar/gni180. PMC 1292996. PMID 16314307.
- Takai D, Jones PA (2002). "Comprehensive analysis of CpG islands in human chromosomes 21 and 22". Proc Natl Acad Sci USA. 99 (6): 3740–5. Bibcode:2002PNAS...99.3740T. doi:10.1073/pnas.052410099. PMC 122594. PMID 11891299.
- Feil R, Berger F (2007). "Convergent evolution of genomic imprinting in plants and mammals". Trends Genet. 23 (4): 192–199. doi:10.1016/j.tig.2007.02.004. PMID 17316885.
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP (2009). "The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores". Nature Genetics. 41 (2): 178–186. doi:10.1038/ng.298. PMC 2729128. PMID 19151715.
- Cohen N, Kenigsberg E, Tanay A (2011). "Primate CpG Islands Are Maintained by Heterogeneous Evolutionary Regimes Involving Minimal Selection". Cell. 145 (5): 773–786. doi:10.1016/j.cell.2011.04.024. PMID 21620139.
- Saxonov S, Berg P, Brutlag DL (2006). "A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters". Proc. Natl. Acad. Sci. U.S.A. 103 (5): 1412–7. Bibcode:2006PNAS..103.1412S. doi:10.1073/pnas.0510310103. PMC 1345710. PMID 16432200.
- Deaton AM, Bird A (2011). "CpG islands and the regulation of transcription". Genes Dev. 25 (10): 1010–22. doi:10.1101/gad.2037511. PMC 3093116. PMID 21576262.
- Chen HY, Shao CJ, Chen FR, Kwan AL, Chen ZP (2010). "Role of ERCC1 promoter hypermethylation in drug resistance to cisplatin in human gliomas". Int. J. Cancer. 126 (8): 1944–54. doi:10.1002/ijc.24772. PMID 19626585.
- Kaur S, Lotsari-Salomaa JE, Seppänen-Kaijansinkko R, Peltomäki P (2016). "MicroRNA Methylation in Colorectal Cancer". Adv. Exp. Med. Biol. Advances in Experimental Medicine and Biology. 937: 109–22. doi:10.1007/978-3-319-42059-2_6. ISBN 978-3-319-42057-8. PMID 27573897.
- Bird A (2002). "DNA methylation patterns and epigenetic memory". Genes Dev. 16 (1): 6–21. doi:10.1101/gad.947102. PMID 11782440.
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (2013). "Cancer genome landscapes". Science. 339 (6127): 1546–58. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
- Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP (2010). "Orphan CpG islands identify numerous conserved promoters in the mammalian genome". PLoS Genet. 6 (9): e1001134. doi:10.1371/journal.pgen.1001134. PMC 2944787. PMID 20885785.
- Wei J, Li G, Dang S, Zhou Y, Zeng K, Liu M (2016). "Discovery and Validation of Hypermethylated Markers for Colorectal Cancer". Dis. Markers. 2016: 1–7. doi:10.1155/2016/2192853. PMC 4963574. PMID 27493446.
- Beggs AD, Jones A, El-Bahrawy M, El-Bahwary M, Abulafi M, Hodgson SV, Tomlinson IP (2013). "Whole-genome methylation analysis of benign and malignant colorectal tumours". J. Pathol. 229 (5): 697–704. doi:10.1002/path.4132. PMC 3619233. PMID 23096130.
- Schnekenburger M, Diederich M (2012). "Epigenetics Offer New Horizons for Colorectal Cancer Prevention". Curr Colorectal Cancer Rep. 8 (1): 66–81. doi:10.1007/s11888-011-0116-z. PMC 3277709. PMID 22389639.
- Friedman RC, Farh KK, Burge CB, Bartel DP (2009). "Most mammalian mRNAs are conserved targets of microRNAs". Genome Res. 19 (1): 92–105. doi:10.1101/gr.082701.108. PMC 2612969. PMID 18955434.
- Rieke DT, Ochsenreither S, Klinghammer K, Seiwert TY, Klauschen F, Tinhofer I, Keilholz U (2016). "Methylation of RAD51B, XRCC3 and other homologous recombination genes is associated with expression of immune checkpoints and an inflammatory signature in squamous cell carcinoma of the head and neck, lung and cervix". Oncotarget. 7 (46): 75379–75393. doi:10.18632/oncotarget.12211. PMC 5342748. PMID 27683114.
- Jin B, Robertson KD (2013). "DNA methyltransferases, DNA damage repair, and cancer". Adv. Exp. Med. Biol. Advances in Experimental Medicine and Biology. 754: 3–29. doi:10.1007/978-1-4419-9967-2_1. ISBN 978-1-4419-9966-5. PMC 3707278. PMID 22956494.
- Muvarak N, Kelley S, Robert C, Baer MR, Perrotti D, Gambacorti-Passerini C, Civin C, Scheibner K, Rassool FV (2015). "c-MYC Generates Repair Errors via Increased Transcription of Alternative-NHEJ Factors, LIG3 and PARP1, in Tyrosine Kinase-Activated Leukemias". Mol. Cancer Res. 13 (4): 699–712. doi:10.1158/1541-7786.MCR-14-0422. PMC 4398615. PMID 25828893.
- Newman EA, Lu F, Bashllari D, Wang L, Opipari AW, Castle VP (2015). "Alternative NHEJ Pathway Components Are Therapeutic Targets in High-Risk Neuroblastoma". Mol. Cancer Res. 13 (3): 470–82. doi:10.1158/1541-7786.MCR-14-0337. PMID 25563294.
- Mego M, Cierna Z, Svetlovska D, Macak D, Machalekova K, Miskovska V, Chovanec M, Usakova V, Obertova J, Babal P, Mardiak J (2013). "PARP expression in germ cell tumours". J. Clin. Pathol. 66 (7): 607–12. doi:10.1136/jclinpath-2012-201088. PMID 23486608.
- Newman RE, Soldatenkov VA, Dritschilo A, Notario V (2002). "Poly(ADP-ribose) polymerase turnover alterations do not contribute to PARP overexpression in Ewing's sarcoma cells". Oncol. Rep. 9 (3): 529–32. doi:10.3892/or.9.3.529. PMID 11956622.
- Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, Kernstine KH, Lin D, Shen B (2008). "Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers". Mol. Cancer Res. 6 (11): 1710–7. doi:10.1158/1541-7786.MCR-08-0269 (inactive 2020-01-22). PMC 2948671. PMID 19010819.
- Lam JS, Seligson DB, Yu H, Li A, Eeva M, Pantuck AJ, Zeng G, Horvath S, Belldegrun AS (2006). "Flap endonuclease 1 is overexpressed in prostate cancer and is associated with a high Gleason score". BJU Int. 98 (2): 445–51. doi:10.1111/j.1464-410X.2006.06224.x. PMID 16879693.
- Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, Song KS, Rho SM, Yoo HS, Yoo HS, Kim YS, Kim JG, Kim NS (2005). "Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells". Clin. Cancer Res. 11 (2 Pt 1): 473–82. PMID 15701830.
- Wang K, Xie C, Chen D (2014). "Flap endonuclease 1 is a promising candidate biomarker in gastric cancer and is involved in cell proliferation and apoptosis". Int. J. Mol. Med. 33 (5): 1268–74. doi:10.3892/ijmm.2014.1682. PMID 24590400.
- Krause A, Combaret V, Iacono I, Lacroix B, Compagnon C, Bergeron C, Valsesia-Wittmann S, Leissner P, Mougin B, Puisieux A (2005). "Genome-wide analysis of gene expression in neuroblastomas detected by mass screening" (PDF). Cancer Lett. 225 (1): 111–20. doi:10.1016/j.canlet.2004.10.035. PMID 15922863.
- Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, van Heek NT, Rosty C, Walter K, Sato N, Parker A, Ashfaq R, Jaffee E, Ryu B, Jones J, Eshleman JR, Yeo CJ, Cameron JL, Kern SE, Hruban RH, Brown PO, Goggins M (2003). "Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays". Am. J. Pathol. 162 (4): 1151–62. doi:10.1016/S0002-9440(10)63911-9. PMC 1851213. PMID 12651607.
- Nikolova T, Christmann M, Kaina B (2009). "FEN1 is overexpressed in testis, lung and brain tumors". Anticancer Res. 29 (7): 2453–9. PMID 19596913.
- Kastan MB (2008). "DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture". Mol. Cancer Res. 6 (4): 517–24. doi:10.1158/1541-7786.MCR-08-0020. PMID 18403632.
- Bernstein, C; Prasad, AR; Nfonsam, V; Bernstein, H. (2013). "Chapter 16: DNA Damage, DNA Repair and Cancer". In Chen, Clark (ed.). New Research Directions in DNA Repair. p. 413. ISBN 978-953-51-1114-6.
- O'Hagan HM, Mohammad HP, Baylin SB (2008). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLoS Genetics. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- Cuozzo C, Porcellini A, Angrisano T, et al. (July 2007). "DNA damage, homology-directed repair, and DNA methylation". PLoS Genetics. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- Horvath S (2013). "DNA methylation age of human tissues and cell types". Genome Biology. 14 (10): R115. doi:10.1186/gb-2013-14-10-r115. PMC 4015143. PMID 24138928.
- Ramirez-Ortiz ZG, Specht CA, Wang JP, Lee CK, Bartholomeu DC, Gazzinelli RT, Levitz SM (2008). "Toll-like receptor 9-dependent immune activation by unmethylated CpG motifs in Aspergillus fumigatus DNA". Infect. Immun. 76 (5): 2123–2129. doi:10.1128/IAI.00047-08. PMC 2346696. PMID 18332208.
- Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, Gnirke A, Meissner A (December 2011). "Genomic distribution and inter-sample variation of non-CpG methylation across human cell types". PLoS Genet. 7 (12): e1002389. doi:10.1371/journal.pgen.1002389. PMC 3234221. PMID 22174693.
- Fasolino M, Zhou Z (May 2017). "The Crucial Role of DNA Methylation and MeCP2 in Neuronal Function". Genes (Basel). 8 (5): 141. doi:10.3390/genes8050141. PMC 5448015. PMID 28505093.
- Duke CG, Kennedy AJ, Gavin CF, Day JJ, Sweatt JD (July 2017). "Experience-dependent epigenomic reorganization in the hippocampus". Learn. Mem. 24 (7): 278–288. doi:10.1101/lm.045112.117. PMC 5473107. PMID 28620075.
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Garcia Vizcaino JC, Schuetz AL, Burkhardt S, Benito E, Navarro Sala M, Javan SB, Haass C, Schmid B, Fischer A, Bonn S (January 2016). "DNA methylation changes in plasticity genes accompany the formation and maintenance of memory". Nat. Neurosci. 19 (1): 102–10. doi:10.1038/nn.4194. PMC 4700510. PMID 26656643.
- Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, Pan F, Zhao J, Hu Z, Sekhar C, Guo Z (September 2016). "OGG1 is essential in oxidative stress induced DNA demethylation". Cell. Signal. 28 (9): 1163–71. doi:10.1016/j.cellsig.2016.05.021. PMID 27251462.
- Bayraktar G, Kreutz MR (2018). "The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders". Front Mol Neurosci. 11: 169. doi:10.3389/fnmol.2018.00169. PMC 5975432. PMID 29875631.
- Massaad CA, Klann E (May 2011). "Reactive oxygen species in the regulation of synaptic plasticity and memory". Antioxid. Redox Signal. 14 (10): 2013–54. doi:10.1089/ars.2010.3208. PMC 3078504. PMID 20649473.
- Beckhauser TF, Francis-Oliveira J, De Pasquale R (2016). "Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity". J Exp Neurosci. 10 (Suppl 1): 23–48. doi:10.4137/JEN.S39887. PMC 5012454. PMID 27625575.
- Day JJ, Sweatt JD (November 2010). "DNA methylation and memory formation". Nat. Neurosci. 13 (11): 1319–23. doi:10.1038/nn.2666. PMC 3130618. PMID 20975755.