Costeff syndrome
Costeff syndrome, or 3-methylglutaconic aciduria type III, is a genetic disorder caused by mutations in the OPA3 gene.[1] It is typically associated with the onset of visual deterioration (optic atrophy) in early childhood followed by the development of movement problems and motor disability in later childhood, occasionally along with mild cases of cognitive deficiency.[2] The disorder is named after Hanan Costeff, the doctor who first described the syndrome in 1989. [3]
| Costeff syndrome | |
|---|---|
| Other names | 3-Methylglutaconic Aciduria Type III, Costeff Optic Atrophy Syndrome, Optic Atrophy Plus Syndrome |
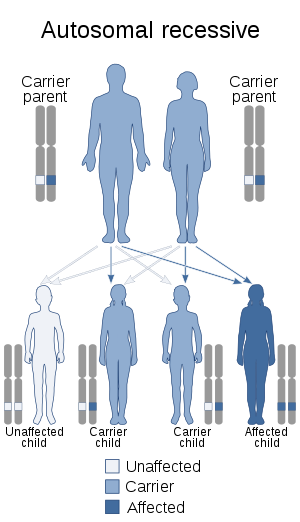 | |
| This condition is inherited in an autosomal recessive manner. | |
Signs and symptoms
The characteristic symptom of Costeff syndrome is the onset of progressively worsening eyesight caused by degeneration of the optic nerve (optic atrophy) within the first few years of childhood, with the majority of affected individuals also developing motor disabilities later in childhood.[4] Occasionally, people with Costeff syndrome may also experience mild cognitive disability.[5]
It is type of 3-methylglutaconic aciduria, the hallmark of which is an increased level in the urinary concentrations of 3-methylglutaconic acid and 3-methylglutaric acid; this can allow diagnosis as early as at one year of age.[2]
Those with Costeff syndrome typically experience the first symptoms of visual deterioration within the first few years of childhood, which manifests as the onset of progressively decreasing visual acuity. This decrease tends to continue with age, even after childhood.[5]
The majority of people with Costeff syndrome develop movement problems and motor disabilities later in childhood, the two most significant of which are choreoathetosis and spasticity.[5] The former causes involuntary erratic, jerky, and twisting movements (see chorea and athetosis), whereas the latter causes twitches and spastic tendencies.
These two symptoms are often severe enough to seriously disable an individual; among 36 people with Costeff syndrome, 17 experienced major motor disability as a result of choreoathetosis, and 12 experienced spasticity-related symptoms severe enough to do the same.[5]
Ataxia (loss of muscle coordination) and speech impairment caused by dysarthria also occur in roughly 50% of cases, but are rarely seriously disabling.[6] Some individuals with Costeff disease also display mild cognitive impairment, though such cases are relatively infrequent.[2]
Genetics
Costeff syndrome is a neuro-ophthalmological genetic disorder; specifically, it is an autosomal recessive disorder caused by a mutation in the OPA3 gene, which carries instructions for synthesis of its gene product, the OPA3 protein. The exact function of the product protein is unknown, though it is known to play an integral role in mitochondrial function.[4][7][8]
The disorder can be caused by several different mutations in the OPA3 gene. However, nearly all reported cases of Costeff syndrome has been in individuals of Iraqi Jewish origin, all of whom share the same splice site founder mutation.[2]
In particular, this founder mutation is a G-to-C mutation of a single nucleotide (see single-nucleotide polymorphism) which interferes with gene expression levels of OPA3.[2] Though the mutation is the same within this population, the severity of symptoms varies with the individual.[5]
To date, there have only been two reported cases of Costeff syndrome not due to the founder mutation, each of which was due to a different pathogenic variants (mutation).[9][10]
Two other OPA3 mutations have also been reported which result in a rare dominant genetic disorder with symptoms similar to Costeff syndrome.[11]
Diagnosis
Costeff syndrome can be diagnosed by the presence of increased levels of 3-methylglutaconic acid in the urine, or 3-methylglutaconic aciduria.[12]
Treatment
There is currently no cure for Costeff syndrome. Treatment is supportive, and thus focuses on management of the symptoms. The resulting visual impairment, spasticity, and movement disorders are treated in the same way as similar cases occurring in the general population.[5]
Prognosis
The long-term prognosis of Costeff syndrome is unknown, though it appears to have no effect on life expectancy at least up to the fourth decade of life.[2] However, as mentioned previously, movement problems can often be severe enough to confine individuals to a wheelchair at an early age, and both visual acuity and spasticity tend to worsen over time.
Epidemiology
First described in 1989, Costeff syndrome has been reported almost exclusively in individuals of Iraqi Jewish origin with only two exceptions, one of whom was a Turkish Kurdish, with the other being of Indian descent.[9][10] Within the Iraqi Jewish population, the carrier frequency of the founder mutation is about 1/10, with the prevalence of Costeff syndrome itself estimated at anywhere between 1 in 400 and 1 in 10,000.[2][3]
See also
References
- Reference, Genetics Home. "Costeff syndrome". Genetics Home Reference. Retrieved 2017-05-28.
- Anikster, Yair; Kleta, Robert; Shaag, Avraham; Gahl, William A.; Elpeleg, Orly (2001–2012). "Type III 3-Methylglutaconic Aciduria (Optic Atrophy Plus Syndrome, or Costeff Optic Atrophy Syndrome): Identification of the OPA3 Gene and Its Founder Mutation in Iraqi Jews". American Journal of Human Genetics. 69 (6): 1218–1224. doi:10.1086/324651. ISSN 0002-9297. PMC 1235533. PMID 11668429.
- Costeff, H.; Gadoth, N.; Apter, N.; Prialnic, M.; Savir, H. (1989–2004). "A familial syndrome of infantile optic atrophy, movement disorder, and spastic paraplegia". Neurology. 39 (4): 595–597. doi:10.1212/wnl.39.4.595. ISSN 0028-3878. PMID 2494568.
- Wortmann, Saskia B.; Kluijtmans, Leo A.; Engelke, Udo F. H.; Wevers, Ron A.; Morava, Eva (January 2012). "The 3-methylglutaconic acidurias: what’s new?". Journal of Inherited Metabolic Disease. 35 (1): 13–22. doi:10.1007/s10545-010-9210-7. ISSN 0141-8955. PMC 3249181. PMID 20882351.
- Gunay-Aygun, Meral; Huizing, Marjan; Anikster, Yair (1993). "OPA3-Related 3-Methylglutaconic Aciduria". In Roberta A. Pagon; Margaret P. Adam; Holly H. Ardinger; et al. (eds.). GeneReviews(®). Seattle (WA): University of Washington, Seattle. PMID 20301646.
- Elpeleg, O. N.; Costeff, H.; Joseph, A.; Shental, Y.; Weitz, R.; Gibson, K. M. (1994–2002). "3-Methylglutaconic aciduria in the Iraqi-Jewish 'optic atrophy plus' (Costeff) syndrome". Developmental Medicine and Child Neurology. 36 (2): 167–172. doi:10.1111/j.1469-8749.1994.tb11825.x. ISSN 0012-1622. PMID 7510656.
- Ryu, Seung-Wook; Jeong, Hyeon Joo; Choi, Myunghwan; Karbowski, Mariusz; Choi, Chulhee (August 2010). "Optic atrophy 3 as a protein of the mitochondrial outer membrane induces mitochondrial fragmentation". Cellular and Molecular Life Sciences. 67 (16): 2839–2850. doi:10.1007/s00018-010-0365-z. ISSN 1420-9071. PMID 20372962.
- Huizing, Marjan; Dorward, Heidi; Ly, Lien; Klootwijk, Enriko; Kleta, Robert; Skovby, Flemming; Pei, Wuhong; Feldman, Benjamin; Gahl, William A.; Anikster, Yair (June 2010). "OPA3, mutated in 3-methylglutaconic aciduria type III, encodes two transcripts targeted primarily to mitochondria". Molecular Genetics and Metabolism. 100 (2): 149–154. doi:10.1016/j.ymgme.2010.03.005. ISSN 1096-7192. PMC 2872056. PMID 20350831.
- Kleta, Robert; Skovby, Flemming; Christensen, Ernst; Rosenberg, Thomas; Gahl, William A.; Anikster, Yair (July 2002). "3-Methylglutaconic aciduria type III in a non-Iraqi-Jewish kindred: clinical and molecular findings". Molecular Genetics and Metabolism. 76 (3): 201–206. doi:10.1016/S1096-7192(02)00047-1. ISSN 1096-7192. PMID 12126933.
- Ho, G.; Walter, J. H.; Christodoulou, J. (December 2008). "Costeff optic atrophy syndrome: new clinical case and novel molecular findings". Journal of Inherited Metabolic Disease. 31 Suppl 2: 419–423. doi:10.1007/s10545-008-0981-z. ISSN 1573-2665. PMID 18985435.
- Reynier, P; Amati-Bonneau, P; Verny, C; Olichon, A; Simard, G; Guichet, A; Bonnemains, C; Malecaze, F; Malinge, M; Pelletier, J; Calvas, P; Dollfus, H; Belenguer, P; Malthiery, Y; Lenaers, G; Bonneau, D (September 2004). "OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract". Journal of Medical Genetics. 41 (9): –110. doi:10.1136/jmg.2003.016576. ISSN 0022-2593. PMC 1735897. PMID 15342707.
- Reference, Genetics Home. "Costeff syndrome". Genetics Home Reference. Retrieved 2019-05-26.
External links
| Classification |
|---|