Circular permutation in proteins
A circular permutation is a relationship between proteins whereby the proteins have a changed order of amino acids in their peptide sequence. The result is a protein structure with different connectivity, but overall similar three-dimensional (3D) shape. In 1979, the first pair of circularly permuted proteins – concanavalin A and lectin – were discovered; over 2000 such proteins are now known.
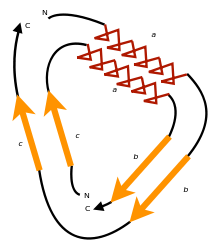
Circular permutation can occur as the result of evolutionary events, posttranslational modifications, or artificially engineered mutations. The two main models proposed to explain the evolution of circularly permuted proteins are permutation by duplication and fission and fusion. Permutation by duplication occurs when a gene undergoes duplication to form a tandem repeat, before redundant sections of the protein are removed; this relationship is found between saposin and swaposin. Fission and fusion occurs when partial proteins fuse to form a single polypeptide, such as in nicotinamide nucleotide transhydrogenases.
Circular permutations are routinely engineered in the laboratory to improve their catalytic activity or thermostability, or to investigate properties of the original protein.
Traditional algorithms for sequence alignment and structure alignment are not able to detect circular permutations between proteins. New non-linear approaches have been developed that overcome this and are able to detect topology-independent similarities.
History
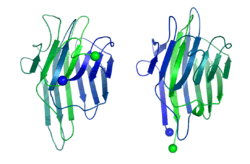
In 1979, Bruce Cunningham and his colleagues discovered the first instance of a circularly permuted protein in nature.[1] After determining the peptide sequence of the lectin protein favin, they noticed its similarity to a known protein – concanavalin A – except that the ends were circularly permuted. Later work confirmed the circular permutation between the pair[2] and showed that concanavalin A is permuted post-translationally[3] through cleavage and an unusual protein ligation.[4]
After the discovery of a natural circularly permuted protein, researchers looked for a way to emulate this process. In 1983, David Goldenberg and Thomas Creighton were able to create a circularly permuted version of a protein by chemically ligating the termini to create a cyclic protein, then introducing new termini elsewhere using trypsin.[5] In 1989, Karolin Luger and her colleagues introduced a genetic method for making circular permutations by carefully fragmenting and ligating DNA.[6] This method allowed for permutations to be introduced at arbitrary sites.[6]
Despite the early discovery of post-translational circular permutations and the suggestion of a possible genetic mechanism for evolving circular permutants, it was not until 1995 that the first circularly permuted pair of genes were discovered. Saposins are a class of proteins involved in sphingolipid catabolism and antigen presentation of lipids in humans. Chris Ponting and Robert Russell identified a circularly permuted version of a saposin inserted into plant aspartic proteinase, which they nicknamed swaposin.[7] Saposin and swaposin were the first known case of two natural genes related by a circular permutation.[7]
Hundreds of examples of protein pairs related by a circular permutation were subsequently discovered in nature or produced in the laboratory. As of February 2012, the Circular Permutation Database[8] contains 2,238 circularly permuted protein pairs with known structures, and many more are known without structures.[9] The CyBase database collects proteins that are cyclic, some of which are permuted variants of cyclic wild-type proteins.[10] SISYPHUS is a database that contains a collection of hand-curated manual alignments of proteins with non-trivial relationships, several of which have circular permutations.[11]
Evolution
There are two main models that are currently being used to explain the evolution of circularly permuted proteins: permutation by duplication and fission and fusion. The two models have compelling examples supporting them, but the relative contribution of each model in evolution is still under debate.[12] Other, less common, mechanisms have been proposed, such as "cut and paste"[13] or "exon shuffling".[14]
Permutation by duplication

The earliest model proposed for the evolution of circular permutations is the permutation by duplication mechanism.[1] In this model, a precursor gene first undergoes a duplication and fusion to form a large tandem repeat. Next, start and stop codons are introduced at corresponding locations in the duplicated gene, removing redundant sections of the protein.
One surprising prediction of the permutation by duplication mechanism is that intermediate permutations can occur. For instance, the duplicated version of the protein should still be functional, since otherwise evolution would quickly select against such proteins. Likewise, partially duplicated intermediates where only one terminus was truncated should be functional. Such intermediates have been extensively documented in protein families such as DNA methyltransferases.[15]
Saposin and swaposin
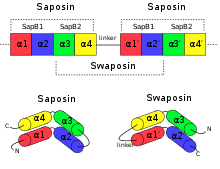
An example for permutation by duplication is the relationship between saposin and swaposin. Saposins are highly conserved glycoproteins, approximately 80 amino acid residues long and forming a four alpha helical structure. They have a nearly identical placement of cysteine residues and glycosylation sites. The cDNA sequence that codes for saposin is called prosaposin. It is a precursor for four cleavage products, the saposins A, B, C, and D. The four saposin domains most likely arose from two tandem duplications of an ancestral gene.[16] This repeat suggests a mechanism for the evolution of the relationship with the plant-specific insert (PSI). The PSI is a domain exclusively found in plants, consisting of approximately 100 residues and found in plant aspartic proteases.[17] It belongs to the saposin-like protein family (SAPLIP) and has the N- and C- termini "swapped", such that the order of helices is 3-4-1-2 compared with saposin, thus leading to the name "swaposin".[7][18]
Fission and fusion
.svg.png)
Another model for the evolution of circular permutations is the fission and fusion model. The process starts with two partial proteins. These may represent two independent polypeptides (such as two parts of a heterodimer), or may have originally been halves of a single protein that underwent a fission event to become two polypeptides.
The two proteins can later fuse together to form a single polypeptide. Regardless of which protein comes first, this fusion protein may show similar function. Thus, if a fusion between two proteins occurs twice in evolution (either between paralogues within the same species or between orthologues in different species) but in a different order, the resulting fusion proteins will be related by a circular permutation.
Evidence for a particular protein having evolved by a fission and fusion mechanism can be provided by observing the halves of the permutation as independent polypeptides in related species, or by demonstrating experimentally that the two halves can function as separate polypeptides.[19]
Transhydrogenases
An example for the fission and fusion mechanism can be found in nicotinamide nucleotide transhydrogenases.[20] These are membrane-bound enzymes that catalyze the transfer of a hydride ion between NAD(H) and NADP(H) in a reaction that is coupled to transmembrane proton translocation. They consist of three major functional units (I, II, and III) that can be found in different arrangement in bacteria, protozoa, and higher eukaryotes. Phylogenetic analysis suggests that the three groups of domain arrangements were acquired and fused independently.[12]
Other processes that can lead to circular permutations
Post-translational modification
The two evolutionary models mentioned above describe ways in which genes may be circularly permuted, resulting in a circularly permuted mRNA after transcription. Proteins can also be circularly permuted via post-translational modification, without permuting the underlying gene. Circular permutations can happen spontaneously through autocatalysis, as in the case of concanavalin A.[4] Alternately, permutation may require restriction enzymes and ligases.[5]
Role in protein engineering
Many proteins have their termini located close together in 3D space.[21][22] Because of this, it is often possible to design circular permutations of proteins. Today, circular permutations are generated routinely in the lab using standard genetics techniques.[6] Although some permutation sites prevent the protein from folding correctly, many permutants have been created with nearly identical structure and function to the original protein.
The motivation for creating a circular permutant of a protein can vary. Scientists may want to improve some property of the protein, such as:
- Reduce proteolytic susceptibility. The rate at which proteins are broken down can have a large impact on their activity in cells. Since termini are often accessible to proteases, designing a circularly permuted protein with less-accessible termini can increase the lifespan of that protein in the cell.[23]
- Improve catalytic activity. Circularly permuting a protein can sometimes increase the rate at which it catalyzes a chemical reaction, leading to more efficient proteins.[24]
- Alter substrate or ligand binding. Circularly permuting a protein can result in the loss of substrate binding, but can occasionally lead to novel ligand binding activity or altered substrate specificity.[25]
- Improve thermostability. Making proteins active over a wider range of temperatures and conditions can improve their utility.[26]
Alternately, scientists may be interested in properties of the original protein, such as:
- Fold order. Determining the order in which different parts of a protein fold is challenging due to the extremely fast time scales involved. Circularly permuted versions of proteins will often fold in a different order, providing information about the folding of the original protein.[27][28][29]
- Essential structural elements. Artificial circularly permuted proteins can allow parts of a protein to be selectively deleted. This gives insight into which structural elements are essential or not.[30]
- Modify quaternary structure. Circularly permuted proteins have been shown to take on different quaternary structure than wild-type proteins.[31]
- Find insertion sites for other proteins. Inserting one protein as a domain into another protein can be useful. For instance, inserting calmodulin into green fluorescent protein (GFP) allowed researchers to measure the activity of calmodulin via the fluorescence of the split-GFP.[32] Regions of GFP that tolerate the introduction of circular permutation are more likely to accept the addition of another protein while retaining the function of both proteins.
- Design of novel biocatalysts and biosensors. Introducing circular permutations can be used to design proteins to catalyze specific chemical reactions,[24][33] or to detect the presence of certain molecules using proteins. For instance, the GFP-calmodulin fusion described above can be used to detect the level of calcium ions in a sample.[32]
Algorithmic detection
Many sequence alignment and protein structure alignment algorithms have been developed assuming linear data representations and as such are not able to detect circular permutations between proteins.[34] Two examples of frequently used methods that have problems correctly aligning proteins related by circular permutation are dynamic programming and many hidden Markov models.[34] As an alternative to these, a number of algorithms are built on top of non-linear approaches and are able to detect topology-independent similarities, or employ modifications allowing them to circumvent the limitations of dynamic programming.[34][35] The table below is a collection of such methods.
The algorithms are classified according to the type of input they require. Sequence-based algorithms require only the sequence of two proteins in order to create an alignment.[36] Sequence methods are generally fast and suitable for searching whole genomes for circularly permuted pairs of proteins.[36] Structure-based methods require 3D structures of both proteins being considered.[37] They are often slower than sequence-based methods, but are able to detect circular permutations between distantly related proteins with low sequence similarity.[37] Some structural methods are topology independent, meaning that they are also able to detect more complex rearrangements than circular permutation.[38]
| NAME | Type | Description | Author | Year | Availability | Reference |
|---|---|---|---|---|---|---|
| FBPLOT | Sequence | Draws dot plots of suboptimal sequence alignments | Zuker | 1991 | [39] | |
| Bachar et al. | Structure, topology independent | Uses geometric hashing for the topology independent comparison of proteins | Bachar et al. | 1993 | [35] | |
| Uliel at al | Sequence | First suggestion of how a sequence comparison algorithm for the detection of circular permutations can work | Uliel et al. | 1999 | [36] | |
| SHEBA | Structure | Uses SHEBA algorithm to create structural alignments for various permutation points, while iteratively improving the cut point. | Jung & Lee | 2001 | [14] | |
| Multiprot | Structure, Topology independent | Calculates a sequence order independent multiple protein structure alignment | Shatsky | 2004 | server, download | [38] |
| RASPODOM | Sequence | Modified Needleman & Wunsch sequence comparison algorithm | Weiner et al. | 2005 | download | [34] |
| CPSARST | Structure | Describes protein structures as one-dimensional text strings by using a Ramachandran sequential transformation (RST) algorithm. Detects circular permutations through a duplication of the sequence representation and "double filter-and-refine" strategy. | Lo, Lyu | 2008 | server | [40] |
| GANGSTA + | Structure | Works in two stages: Stage one identifies coarse alignments based on secondary structure elements. Stage two refines the alignment on residue level and extends into loop regions. | Schmidt-Goenner et al. | 2009 | server, download | [41] |
| SANA | Structure | Detect initial aligned fragment pairs (AFPs). Build network of possible AFPs. Use random-mate algorithm to connect components to a graph. | Wang et al. | 2010 | download | [42] |
| CE-CP | Structure | Built on top of the combinatorial extension algorithm. Duplicates atoms before alignment, truncates results after alignment | Bliven et al. | 2015 | server, download | [43] |
| TopMatch | Structure | Has option to calculate topology-independent protein structure alignment | Sippl & Wiederstein | 2012 | server, download | [44] |
References
![]()
- Cunningham BA, Hemperly JJ, Hopp TP, Edelman GM (July 1979). "Favin versus concanavalin A: Circularly permuted amino acid sequences". Proceedings of the National Academy of Sciences of the United States of America. 76 (7): 3218–22. doi:10.1073/pnas.76.7.3218. PMC 383795. PMID 16592676.
- Einspahr H, Parks EH, Suguna K, Subramanian E, Suddath FL (December 1986). "The crystal structure of pea lectin at 3.0-A resolution". The Journal of Biological Chemistry. 261 (35): 16518–27. PMID 3782132.
- Carrington DM, Auffret A, Hanke DE (1985). "Polypeptide ligation occurs during post-translational modification of concanavalin A". Nature. 313 (5997): 64–7. doi:10.1038/313064a0. PMID 3965973.
- Bowles DJ, Pappin DJ (February 1988). "Traffic and assembly of concanavalin A". Trends in Biochemical Sciences. 13 (2): 60–4. doi:10.1016/0968-0004(88)90030-8. PMID 3070848.
- Goldenberg DP, Creighton TE (April 1983). "Circular and circularly permuted forms of bovine pancreatic trypsin inhibitor". Journal of Molecular Biology. 165 (2): 407–13. doi:10.1016/S0022-2836(83)80265-4. PMID 6188846.
- Luger K, Hommel U, Herold M, Hofsteenge J, Kirschner K (January 1989). "Correct folding of circularly permuted variants of a beta alpha barrel enzyme in vivo". Science. 243 (4888): 206–10. doi:10.1126/science.2643160. PMID 2643160.
- Ponting CP, Russell RB (May 1995). "Swaposins: circular permutations within genes encoding saposin homologues". Trends in Biochemical Sciences. 20 (5): 179–80. doi:10.1016/S0968-0004(00)89003-9. PMID 7610480.
- Lo W, Lee C, Lee C, Lyu P. "Circular Permutation Database". Institute of Bioinformatics and Structural Biology, National Tsing Hua University. Retrieved 16 February 2012.
- Lo WC, Lee CC, Lee CY, Lyu PC (January 2009). "CPDB: a database of circular permutation in proteins". Nucleic Acids Research. 37 (Database issue): D328–32. doi:10.1093/nar/gkn679. PMC 2686539. PMID 18842637.
- Kaas Q, Craik DJ (2010). "Analysis and classification of circular proteins in CyBase". Biopolymers. 94 (5): 584–91. doi:10.1002/bip.21424. PMID 20564021.
- Andreeva A, Prlić A, Hubbard TJ, Murzin AG (January 2007). "SISYPHUS--structural alignments for proteins with non-trivial relationships". Nucleic Acids Research. 35 (Database issue): D253–9. doi:10.1093/nar/gkl746. PMC 1635320. PMID 17068077.
- Weiner J, Bornberg-Bauer E (April 2006). "Evolution of circular permutations in multidomain proteins". Molecular Biology and Evolution. 23 (4): 734–43. doi:10.1093/molbev/msj091. PMID 16431849.
- Bujnicki JM (March 2002). "Sequence permutations in the molecular evolution of DNA methyltransferases". BMC Evolutionary Biology. 2 (1): 3. doi:10.1186/1471-2148-2-3. PMC 102321. PMID 11914127.
- Jung J, Lee B (September 2001). "Circularly permuted proteins in the protein structure database". Protein Science. 10 (9): 1881–6. doi:10.1110/ps.05801. PMC 2253204. PMID 11514678.
- Jeltsch A (July 1999). "Circular permutations in the molecular evolution of DNA methyltransferases". Journal of Molecular Evolution. 49 (1): 161–4. doi:10.1007/pl00006529. PMID 10368444.
- Hazkani-Covo E, Altman N, Horowitz M, Graur D (January 2002). "The evolutionary history of prosaposin: two successive tandem-duplication events gave rise to the four saposin domains in vertebrates". Journal of Molecular Evolution. 54 (1): 30–4. doi:10.1007/s00239-001-0014-0. PMID 11734895.
- Guruprasad K, Törmäkangas K, Kervinen J, Blundell TL (September 1994). "Comparative modelling of barley-grain aspartic proteinase: a structural rationale for observed hydrolytic specificity". FEBS Letters. 352 (2): 131–6. doi:10.1016/0014-5793(94)00935-X. PMID 7925961.
- Bruhn H (July 2005). "A short guided tour through functional and structural features of saposin-like proteins". The Biochemical Journal. 389 (Pt 2): 249–57. doi:10.1042/BJ20050051. PMC 1175101. PMID 15992358.
- Lee J, Blaber M (January 2011). "Experimental support for the evolution of symmetric protein architecture from a simple peptide motif". Proceedings of the National Academy of Sciences of the United States of America. 108 (1): 126–30. doi:10.1073/pnas.1015032108. PMC 3017207. PMID 21173271.
- Hatefi Y, Yamaguchi M (March 1996). "Nicotinamide nucleotide transhydrogenase: a model for utilization of substrate binding energy for proton translocation". FASEB Journal. 10 (4): 444–52. doi:10.1096/fasebj.10.4.8647343. PMID 8647343.
- Thornton JM, Sibanda BL (June 1983). "Amino and carboxy-terminal regions in globular proteins". Journal of Molecular Biology. 167 (2): 443–60. doi:10.1016/S0022-2836(83)80344-1. PMID 6864804.
- Yu Y, Lutz S (January 2011). "Circular permutation: a different way to engineer enzyme structure and function". Trends in Biotechnology. 29 (1): 18–25. doi:10.1016/j.tibtech.2010.10.004. PMID 21087800.
- Whitehead TA, Bergeron LM, Clark DS (October 2009). "Tying up the loose ends: circular permutation decreases the proteolytic susceptibility of recombinant proteins". Protein Engineering, Design & Selection. 22 (10): 607–13. doi:10.1093/protein/gzp034. PMID 19622546.
- Cheltsov AV, Barber MJ, Ferreira GC (June 2001). "Circular permutation of 5-aminolevulinate synthase. Mapping the polypeptide chain to its function". The Journal of Biological Chemistry. 276 (22): 19141–9. doi:10.1074/jbc.M100329200. PMC 4547487. PMID 11279050.
- Qian Z, Lutz S (October 2005). "Improving the catalytic activity of Candida antarctica lipase B by circular permutation". Journal of the American Chemical Society. 127 (39): 13466–7. doi:10.1021/ja053932h. PMID 16190688. (primary source)
- Topell S, Hennecke J, Glockshuber R (August 1999). "Circularly permuted variants of the green fluorescent protein". FEBS Letters. 457 (2): 283–9. doi:10.1016/S0014-5793(99)01044-3. PMID 10471794. (primary source)
- Viguera AR, Serrano L, Wilmanns M (October 1996). "Different folding transition states may result in the same native structure". Nature Structural Biology. 3 (10): 874–80. doi:10.1038/nsb1096-874. PMID 8836105. (primary source)
- Capraro DT, Roy M, Onuchic JN, Jennings PA (September 2008). "Backtracking on the folding landscape of the beta-trefoil protein interleukin-1beta?". Proceedings of the National Academy of Sciences of the United States of America. 105 (39): 14844–8. doi:10.1073/pnas.0807812105. PMC 2567455. PMID 18806223.
- Zhang P, Schachman HK (July 1996). "In vivo formation of allosteric aspartate transcarbamoylase containing circularly permuted catalytic polypeptide chains: implications for protein folding and assembly". Protein Science. 5 (7): 1290–300. doi:10.1002/pro.5560050708. PMC 2143468. PMID 8819162. (primary source)
- Huang YM, Nayak S, Bystroff C (November 2011). "Quantitative in vivo solubility and reconstitution of truncated circular permutants of green fluorescent protein". Protein Science. 20 (11): 1775–80. doi:10.1002/pro.735. PMC 3267941. PMID 21910151. (primary source)
- Beernink PT, Yang YR, Graf R, King DS, Shah SS, Schachman HK (March 2001). "Random circular permutation leading to chain disruption within and near alpha helices in the catalytic chains of aspartate transcarbamoylase: effects on assembly, stability, and function". Protein Science. 10 (3): 528–37. doi:10.1110/ps.39001. PMC 2374132. PMID 11344321.
- Baird GS, Zacharias DA, Tsien RY (September 1999). "Circular permutation and receptor insertion within green fluorescent proteins". Proceedings of the National Academy of Sciences of the United States of America. 96 (20): 11241–6. doi:10.1073/pnas.96.20.11241. PMC 18018. PMID 10500161.
- Turner NJ (August 2009). "Directed evolution drives the next generation of biocatalysts". Nature Chemical Biology. 5 (8): 567–73. doi:10.1038/nchembio.203. PMID 19620998.
- Weiner J, Thomas G, Bornberg-Bauer E (April 2005). "Rapid motif-based prediction of circular permutations in multi-domain proteins". Bioinformatics. 21 (7): 932–7. doi:10.1093/bioinformatics/bti085. PMID 15788783.
- Bachar O, Fischer D, Nussinov R, Wolfson H (April 1993). "A computer vision based technique for 3-D sequence-independent structural comparison of proteins". Protein Engineering. 6 (3): 279–88. doi:10.1093/protein/6.3.279. PMID 8506262.
- Uliel S, Fliess A, Amir A, Unger R (November 1999). "A simple algorithm for detecting circular permutations in proteins". Bioinformatics. 15 (11): 930–6. doi:10.1093/bioinformatics/15.11.930. PMID 10743559.
- Prlic A, Bliven S, Rose PW, Bluhm WF, Bizon C, Godzik A, Bourne PE (December 2010). "Pre-calculated protein structure alignments at the RCSB PDB website". Bioinformatics. 26 (23): 2983–5. doi:10.1093/bioinformatics/btq572. PMC 3003546. PMID 20937596.
- Shatsky M, Nussinov R, Wolfson HJ (July 2004). "A method for simultaneous alignment of multiple protein structures". Proteins. 56 (1): 143–56. doi:10.1002/prot.10628. PMID 15162494.
- Zuker M (September 1991). "Suboptimal sequence alignment in molecular biology. Alignment with error analysis". Journal of Molecular Biology. 221 (2): 403–20. doi:10.1016/0022-2836(91)80062-Y. PMID 1920426.
- Lo WC, Lyu PC (January 2008). "CPSARST: an efficient circular permutation search tool applied to the detection of novel protein structural relationships". Genome Biology. 9 (1): R11. doi:10.1186/gb-2008-9-1-r11. PMC 2395249. PMID 18201387.
- Schmidt-Goenner T, Guerler A, Kolbeck B, Knapp EW (May 2010). "Circular permuted proteins in the universe of protein folds". Proteins. 78 (7): 1618–30. doi:10.1002/prot.22678. PMID 20112421.
- Wang L, Wu LY, Wang Y, Zhang XS, Chen L (July 2010). "SANA: an algorithm for sequential and non-sequential protein structure alignment". Amino Acids. 39 (2): 417–25. doi:10.1007/s00726-009-0457-y. PMID 20127263.
- Bliven SE, Bourne PE, Prlić A (April 2015). "Detection of circular permutations within protein structures using CE-CP". Bioinformatics. 31 (8): 1316–8. doi:10.1093/bioinformatics/btu823. PMC 4393524. PMID 25505094.
- Sippl MJ, Wiederstein M (April 2012). "Detection of spatial correlations in protein structures and molecular complexes". Structure. 20 (4): 718–28. doi:10.1016/j.str.2012.01.024. PMC 3320710. PMID 22483118.
Further reading
- David Goodsell (April 2010) Concanavalin A and Circular Permutation Protein Data Bank (PDB) Molecule of the Month