ChIP-on-chip
ChIP-on-chip (also known as ChIP-chip) is a technology that combines chromatin immunoprecipitation ('ChIP') with DNA microarray ("chip"). Like regular ChIP, ChIP-on-chip is used to investigate interactions between proteins and DNA in vivo. Specifically, it allows the identification of the cistrome, the sum of binding sites, for DNA-binding proteins on a genome-wide basis.[1] Whole-genome analysis can be performed to determine the locations of binding sites for almost any protein of interest.[1] As the name of the technique suggests, such proteins are generally those operating in the context of chromatin. The most prominent representatives of this class are transcription factors, replication-related proteins, like origin recognition complex protein (ORC), histones, their variants, and histone modifications.
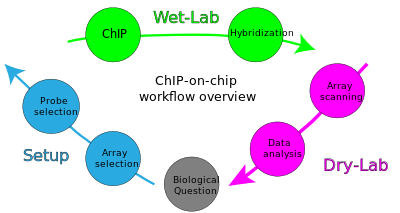
The goal of ChIP-on-chip is to locate protein binding sites that may help identify functional elements in the genome. For example, in the case of a transcription factor as a protein of interest, one can determine its transcription factor binding sites throughout the genome. Other proteins allow the identification of promoter regions, enhancers, repressors and silencing elements, insulators, boundary elements, and sequences that control DNA replication.[2] If histones are subject of interest, it is believed that the distribution of modifications and their localizations may offer new insights into the mechanisms of regulation.
One of the long-term goals ChIP-on-chip was designed for is to establish a catalogue of (selected) organisms that lists all protein-DNA interactions under various physiological conditions. This knowledge would ultimately help in the understanding of the machinery behind gene regulation, cell proliferation, and disease progression. Hence, ChIP-on-chip offers both potential to complement our knowledge about the orchestration of the genome on the nucleotide level and information on higher levels of information and regulation as it is propagated by research on epigenetics.
Technological platforms
The technical platforms to conduct ChIP-on-chip experiments are DNA microarrays, or "chips". They can be classified and distinguished according to various characteristics:
Probe type: DNA arrays can comprise either mechanically spotted cDNAs or PCR-products, mechanically spotted oligonucleotides, or oligonucleotides that are synthesized in situ. The early versions of microarrays were designed to detect RNAs from expressed genomic regions (open reading frames aka ORFs). Although such arrays are perfectly suited to study gene expression profiles, they have limited importance in ChIP experiments since most "interesting" proteins with respect to this technique bind in intergenic regions. Nowadays, even custom-made arrays can be designed and fine-tuned to match the requirements of an experiment. Also, any sequence of nucleotides can be synthesized to cover genic as well as intergenic regions.
Probe size: Early version of cDNA arrays had a probe length of about 200bp. Latest array versions use oligos as short as 70- (Microarrays, Inc.) to 25-mers (Affymetrix). (Feb 2007)
Probe composition: There are tiled and non-tiled DNA arrays. Non-tiled arrays use probes selected according to non-spatial criteria, i.e., the DNA sequences used as probes have no fixed distances in the genome. Tiled arrays, however, select a genomic region (or even a whole genome) and divide it into equal chunks. Such a region is called tiled path. The average distance between each pair of neighboring chunks (measured from the center of each chunk) gives the resolution of the tiled path. A path can be overlapping, end-to-end or spaced.[3]
Array size: The first microarrays used for ChIP-on-Chip contained about 13,000 spotted DNA segments representing all ORFs and intergenic regions from the yeast genome.[2] Nowadays, Affymetrix offers whole-genome tiled yeast arrays with a resolution of 5bp (all in all 3.2 million probes). Tiled arrays for the human genome become more and more powerful, too. Just to name one example, Affymetrix offers a set of seven arrays with about 90 million probes, spanning the complete non-repetitive part of the human genome with about 35bp spacing. (Feb 2007) Besides the actual microarray, other hard- and software equipment is necessary to run ChIP-on-chip experiments. It is generally the case that one company’s microarrays can not be analyzed by another company’s processing hardware. Hence, buying an array requires also buying the associated workflow equipment. The most important elements are, among others, hybridization ovens, chip scanners, and software packages for subsequent numerical analysis of the raw data.
Workflow of a ChIP-on-chip experiment
Starting with a biological question, a ChIP-on-chip experiment can be divided into three major steps: The first is to set up and design the experiment by selecting the appropriate array and probe type. Second, the actual experiment is performed in the wet-lab. Last, during the dry-lab portion of the cycle, gathered data are analyzed to either answer the initial question or lead to new questions so that the cycle can start again.
Wet-lab portion of the workflow
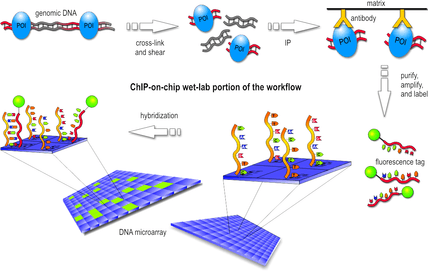
In the first step, the protein of interest (POI) is cross-linked with the DNA site it binds to in an in vitro environment. Usually this is done by a gentle formaldehyde fixation that is reversible with heat.
Then, the cells are lysed and the DNA is sheared by sonication or using micrococcal nuclease. This results in double-stranded chunks of DNA fragments, normally 1 kb or less in length. Those that were cross-linked to the POI form a POI-DNA complex.
In the next step, only these complexes are filtered out of the set of DNA fragments, using an antibody specific to the POI. The antibodies may be attached to a solid surface, may have a magnetic bead, or some other physical property that allows separation of cross-linked complexes and unbound fragments. This procedure is essentially an immunoprecipitation (IP) of the protein. This can be done either by using a tagged protein with an antibody against the tag (ex. FLAG, HA, c-myc) or with an antibody to the native protein.
The cross-linking of POI-DNA complexes is reversed (usually by heating) and the DNA strands are purified. For the rest of the workflow, the POI is no longer necessary.
After an amplification and denaturation step, the single-stranded DNA fragments are labeled with a fluorescent tag such as Cy5 or Alexa 647.
Finally, the fragments are poured over the surface of the DNA microarray, which is spotted with short, single-stranded sequences that cover the genomic portion of interest. Whenever a labeled fragment "finds" a complementary fragment on the array, they will hybridize and form again a double-stranded DNA fragment.
Dry-lab portion of the workflow
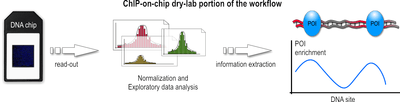
After a sufficiently large time frame to allow hybridization, the array is illuminated with fluorescent light. Those probes on the array that are hybridized to one of the labeled fragments emit a light signal that is captured by a camera. This image contains all raw data for the remaining part of the workflow.
This raw data, encoded as false-color image, needs to be converted to numerical values before the actual analysis can be done. The analysis and information extraction of the raw data often remains the most challenging part for ChIP-on-chip experiments. Problems arise throughout this portion of the workflow, ranging from the initial chip read-out, to suitable methods to subtract background noise, and finally to appropriate algorithms that normalize the data and make it available for subsequent statistical analysis, which then hopefully lead to a better understanding of the biological question that the experiment seeks to address. Furthermore, due to the different array platforms and lack of standardization between them, data storage and exchange is a huge problem. Generally speaking, the data analysis can be divided into three major steps:
During the first step, the captured fluorescence signals from the array are normalized, using control signals derived from the same or a second chip. Such control signals tell which probes on the array were hybridized correctly and which bound nonspecifically.
In the second step, numerical and statistical tests are applied to control data and IP fraction data to identify POI-enriched regions along the genome. The following three methods are used widely: median percentile rank, single-array error, and sliding-window. These methods generally differ in how low-intensity signals are handled, how much background noise is accepted, and which trait for the data is emphasized during the computation. In the recent past, the sliding-window approach seems to be favored and is often described as most powerful.
In the third step, these regions are analyzed further. If, for example, the POI was a transcription factor, such regions would represent its binding sites. Subsequent analysis then may want to infer nucleotide motifs and other patterns to allow functional annotation of the genome.[4]
Strengths and weaknesses
Using tiled arrays, ChIP-on-chip allows for high resolution of genome-wide maps. These maps can determine the binding sites of many DNA-binding proteins like transcription factors and also chromatin modifications.
Although ChIP-on-chip can be a powerful technique in the area of genomics, it is very expensive. Most published studies using ChIP-on-chip repeat their experiments at least three times to ensure biologically meaningful maps. The cost of the DNA microarrays is often a limiting factor to whether a laboratory should proceed with a ChIP-on-chip experiment. Another limitation is the size of DNA fragments that can be achieved. Most ChIP-on-chip protocols utilize sonication as a method of breaking up DNA into small pieces. However, sonication is limited to a minimal fragment size of 200 bp. For higher resolution maps, this limitation should be overcome to achieve smaller fragments, preferably to single nucleosome resolution. As mentioned previously, the statistical analysis of the huge amount of data generated from arrays is a challenge and normalization procedures should aim to minimize artifacts and determine what is really biologically significant. So far, application to mammalian genomes has been a major limitation, for example, due to the significant percentage of the genome that is occupied by repeats. However, as ChIP-on-chip technology advances, high resolution whole mammalian genome maps should become achievable.
Antibodies used for ChIP-on-chip can be an important limiting factor. ChIP-on-chip requires highly specific antibodies that must recognize its epitope in free solution and also under fixed conditions. If it is demonstrated to successfully immunoprecipitate cross-linked chromatin, it is termed "ChIP-grade". Companies that provide ChIP-grade antibodies include Abcam, Cell Signaling Technology, Santa Cruz, and Upstate. To overcome the problem of specificity, the protein of interest can be fused to a tag like FLAG or HA that are recognized by antibodies. An alternative to ChIP-on-chip that does not require antibodies is DamID.
Also available are antibodies against a specific histone modification like H3 tri methyl K4. As mentioned before, the combination of these antibodies and ChIP-on-chip has become extremely powerful in determining whole genome analysis of histone modification patterns and will contribute tremendously to our understanding of the histone code and epigenetics.
A study demonstrating the non-specific nature of DNA binding proteins has been published in PLoS Biology. This indicates that alternate confirmation of functional relevancy is a necessary step in any ChIP-chip experiment.[5]
History
A first ChIP-on-chip experiment was performed in 1999 to analyze the distribution of cohesin along budding yeast chromosome III.[6] Although the genome was not completely represented, the protocol in this study remains equivalent as those used in later studies. The ChIP-on-chip technique using all of the ORFs of the genome (that nevertheless remains incomplete, missing intergenic regions) was then applied successfully in three papers published in 2000 and 2001.[7][8][9] The authors identified binding sites for individual transcription factors in the budding yeast Saccharomyces cerevisiae. In 2002, Richard Young’s group[10] determined the genome-wide positions of 106 transcription factors using a c-Myc tagging system in yeast. The first demonstration of the mammalian ChIp-on-chip technique reported the isolation of nine chromatin fragments containing weak and strong E2F binding site was done by Peggy Farnham's lab in collaboration with Michael Zhang's lab and published in 2001.[11] This study was followed several months later in a collaboration between the Young lab with the laboratory of Brian Dynlacht which used the ChIP-on-chip technique to show for the first time that E2F targets encode components of the DNA damage checkpoint and repair pathways, as well as factors involved in chromatin assembly/condensation, chromosome segregation, and the mitotic spindle checkpoint[12] Other applications for ChIP-on-chip include DNA replication, recombination, and chromatin structure. Since then, ChIP-on-chip has become a powerful tool in determining genome-wide maps of histone modifications and many more transcription factors. ChIP-on-chip in mammalian systems has been difficult due to the large and repetitive genomes. Thus, many studies in mammalian cells have focused on select promoter regions that are predicted to bind transcription factors and have not analyzed the entire genome. However, whole mammalian genome arrays have recently become commercially available from companies like Nimblegen. In the future, as ChIP-on-chip arrays become more and more advanced, high resolution whole genome maps of DNA-binding proteins and chromatin components for mammals will be analyzed in more detail.
Alternatives
Chip-Sequencing is a recently developed technology that still uses chromatin immunoprecipitation to crosslink the proteins of interest to the DNA but then instead of using a micro-array, it uses the more accurate, higher throughput method of sequencing to localize interaction points.
DamID is an alternative method that does not require antibodies.
ChIP-exo uses exonuclease treatment to achieve up to single base pair resolution.
CUT&RUN sequencing uses antibody recognition with targeted enzymatic cleavage to address some technical limitations of ChIP
References
- Aparicio, O; Geisberg, JV; Struhl, K (2004). Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Current Protocols in Cell Biology. Chapter 17. University of Southern California, Los Angeles, California, USA: John Wiley & Sons, Inc. pp. Unit 17.7. doi:10.1002/0471143030.cb1707s23. ISBN 978-0-471-14303-1. ISSN 1934-2616. PMID 18228445.
- M.J. Buck, J.D. Lieb, ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments, Genomics 83 (2004) 349-360.
- Royce TE, Rozowsky JS, Bertone P, Samanta M, Stolc V, Weissman S, Snyder M, Gerstein M. Related Articles. Issues in the analysis of oligonucleotide tiling microarrays for transcript mapping. Trends Genet. 2005 Aug;21(8):466-75. Review.
- "Biomedical Genomics Core - The Research Institute at Nationwide Children's Hospital". genomics.nchresearch.org.
- Li, Xiao-Yong; MacArthur, Stewart; Bourgon, Richard; Nix, David; Pollard, Daniel A.; Iyer, Venky N.; Hechmer, Aaron; Simirenko, Lisa; Stapleton, Mark; Hendriks, Cris L. Luengo; Chu, Hou Cheng; Ogawa, Nobuo; Inwood, William; Sementchenko, Victor; Beaton, Amy; Weiszmann, Richard; Celniker, Susan E.; Knowles, David W.; Gingeras, Tom; Speed, Terence P.; Eisen, Michael B.; Biggin, Mark D. (2008). "Transcription Factors Bind Thousands of Active and Inactive Regions in the Drosophila Blastoderm". PLOS Biology. 6 (2): e27. doi:10.1371/journal.pbio.0060027. PMC 2235902. PMID 18271625.
- Blat Y, Kleckner N., Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region, Cell (1999) Jul 23;98(2):249-59.
- J.D. Lieb, X. Liu, D. Botstein, P.O. Brown, Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association, Nat. Genet. 28 (2001) 327-334.
- B. Ren, F. Robert, J.J. Wyrick, O. Aparicio, E.G. Jennings, I. Simon, J. Zeitlinger, J. Schreiber, N. Nannett, E. Kanin, T.L. Volkert, C.J. Wilson, S.R. Bell, R.A. Young, Genome-wide location and function of DNA binding proteins, Science 290 (2000) 2306-2309.
- V.R. Iyer, C.E. Horak, C.S. Scafe, D. Botstein, M. Snyder, P.O. Brown, Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF, Nature 409 (2001) 533-538.
- T.I. Lee, N.J. Rinaldi, F. Robert, D.T. Odom, Z. Bar-Joseph, G.K. Gerber, N.M. Hannett, C.T. Harbison, C.M. Thompson, I. Simon, J. Zeitlinger, E.G. Jennings, H.L. Murray, D.B. Gordon, B. Ren, J.J. Wyrick, J.B Tagne, T.L. Volkert, E. Fraenkel, D.K. Gifford, R.A Young, Transcriptional regulatory networks in Saccharomyces cerevisiae, Science 298 (2002) 799-804.
- Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Use of chromatin immunoprecipation to clone novel E2F target promoters., Molecular and Cell Biology 20 (2001)6820-32.
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell cycle progression with DNA repair, and G2(M) checkpoints., Genes and Development 16 (2002) 245-56.
Further reading
- Johnson, W. E.; Li, W.; Meyer, C. A.; Gottardo, R.; Carroll, J. S.; Brown, M.; Liu, X. S. (2006). "Model-based analysis of tiling-arrays for ChIP-chip". Proceedings of the National Academy of Sciences. 103 (33): 12457–12462. Bibcode:2006PNAS..10312457J. doi:10.1073/pnas.0601180103. ISSN 0027-8424. PMC 1567901. PMID 16895995.
- Benoukraf, Touati; Cauchy, Pierre; Fenouil, Romain; Jeanniard, Adrien; Koch, Frederic; Jaeger, Sébastien; Thieffry, Denis; Imbert, Jean; Andrau, Jean-Christophe; Spicuglia, Salvatore; Ferrier, Pierre (2009). "CoCAS: a ChIP-on-chip analysis suite". Bioinformatics. 25 (7): 954–955. doi:10.1093/bioinformatics/btp075. ISSN 1460-2059. PMC 2660873. PMID 19193731.
External links
- http://www.genome.gov/10005107 ENCODE project
- Chip-on-Chip (CoC) Package Information from Amkor Technology