Asthma-related microbes
Chronic Mycoplasma pneumonia and Chlamydia pneumonia infections are associated with the onset and exacerbation of asthma.[1] These microbial infections result in chronic lower airway inflammation, impaired mucociliary clearance, an increase in mucous production and eventually asthma. Furthermore, children who experience severe viral respiratory infections early in life have a high possibility of having asthma later in their childhood. These viral respiratory infections are mostly caused by respiratory syncytial virus (RSV) and human rhinovirus (HRV). Although RSV infections increase the risk of asthma in early childhood, the association between asthma and RSV decreases with increasing age. HRV on the other hand is an important cause of bronchiolitis and is strongly associated with asthma development. In children and adults with established asthma, viral upper respiratory tract infections (URIs), especially HRVs infections, can produce acute exacerbations of asthma. Thus, Chlamydia pneumoniae, Mycoplasma pneumoniae and human rhinoviruses are microbes that play a major role in non-atopic asthma.[1]
Asthma
According to Hertzen (2002), a common characteristic of asthmatic patients is to have epithelial cells that respond to injury by enhancing the capability of producing proinflammatory and profibrogenic cytokines, instead of repairing the injured epithelial layer.[2] As a result, inflammation and associated healing process leads to scar formation and tissue remodelling, which are symptoms that can be found in almost all asthmatics patients. Thus, asthma is a chronic inflammatory disorder of the airways. Asthma is divided into two subgroups: atopic (extrinsic) and non-atopic (intrinsic). The atopic subgroup is closely associated with family history of the disease, whereas the non-atopic subgroup has its onset in adulthood and it is not caused by inheritance. It is known that non-atopic asthma has a more severe clinical course than atopic asthma. Non-atopic asthma may be caused by chronic viral, bacterial infections, or colonization with pathogenic bacteria.[2]
Associated Microorganisms
Chlamydophila pneumoniae
General descriptions
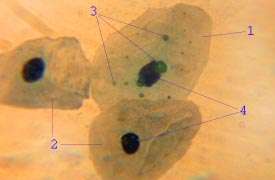
Chlamydophila pneumoniae, formerly known as Chlamydia pneumoniae, is a bacterium that belongs to the phylum Chlamydiae, order Chlamydiales, and genus Chlamydophila.[3] It is rod-shaped and Gram-negative.[3] It has a characteristic pear-shaped elementary body (EB) that is surrounded by a periplasmic space, which makes it morphologically distinct from the round EBs of C. trachomatis and C. psittaci.[4] C. pneumoniae is non-motile and utilizes aerobic respiration. As an obligate intracellular bacterium, C. pneumoniae is both parasitic and mesophilic.[4]
Biological interactions with host
C. pneumoniae is able to grow in monocytes, macrophages, endothelial and smooth muscle cells.[2] It replicates within the host cell cytoplasm. Due to the fact that it does not have the ability to synthesize its own ATP, it is entirely dependent on energy produced by their host.[2][5] Reinfection of the host with C. pneumoniae is common because the memory immunity elicited by C. pneumoniae is short-lived and partial.[2] In addition, C. pneumoniae infection tends to be persistence due to IFN-γ, penicillin and nutrient deficiencies.[6] These deficiencies prevent C.pneumoniae from completing their normal developmental cycle, leading to the formation of aberrant, noninfectious C. pneumoniae that persist in the human host.[6] C. pneumonia infection may not only be persistent and chronic, but it also has irreversible tissue injury and scarring processes,[2] which are symptoms in asthma patients. Infection with C. pneumoniae induces both humoral and cell-mediated immune responses.[2] Among the two immune responses, cell-mediated immune response that involves CD8+ T cells in particular is crucial to eradicate C. pneumoniae, whereas the humoral immune response appears to be rather ineffective in protection against C. pneumonia infection.[2] In fact, CD8+ T cells are so important that if it is absent in the host, the C. pneumonia infection would progress rapidly. Although cell-mediated immune response is responsible for the clearance of C. pneumoniae, this response can be harmful to the host because it favours the development of inflammation that can lead to asthma.[2]
Roles in asthma
There is a strong association of C. pneumoniae with long-standing asthma among the non-atopic asthma in comparison to atopic asthma.[2] In fact, the severity of asthma can be determined by the elevated titres to C. pneumoniae, but not to other potential pathogens such as Mycoplasma pneumoniae, adenovirus, influenza A and B or parainfluenza virus.[2] It is hypothesized that C. pneumoniae is associated with asthma because C. pneumoniae has been found to cause ciliostasis in bronchial epithelial cells.[2] Meanwhile, sero-epidemiological data also provide evidences to support that C. pneumoniae plays a role in asthma by amplifying inflammation and inciting the disease process.[2] The association of C. pneumoniae and asthma begins with C. pneumoniae producing 60-kDa heat shock proteins that provide a prolonged antigenic stimulation.[2] This particular heat shock protein is known as a member of hsp60 family of stress proteins, which can be found in both eukaryotes and prokaryotes. The production of hsp60 remains unaltered even when C. pneumoniae is dormant and does not replicate, since that hsp60 serves as a protective antigen.[2] Its antigenic stimulation strongly amplifies chronic inflammation by increasing the production of proinflammatory cytokines, tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and IFN-γ by infected cells, which ultimately leads to immunopathological tissue damage and scarring in the asthmatic lungs.[2] Furthermore, infection with C. pneumonia also induces serum immunoglobulin M (IgM), IgA, and IgG responses, which are associated with chronic asthma.[4]
Mycoplasma pneumoniae
General descriptions
M. pneumoniae is a bacterium that belongs to the phylum Firmicutes, class Mollicutes, order Mycoplasmatales and family Mycoplasmataceae.[7] It is either filamentous or spherical. Individual spindle-shaped cells of M. pneumoniae are 1 to 2 µm long and 0.1 to 0.2 µm wide.[7] M. pneumoniae is a motile, mesophilic bacterium that exhibits obligate aerobic respiration. It is an extracellular, host-associated bacterium that lacks a cell wall[8] and is unable to survive outside of a host due to osmotic instability in the environment.
Biological interactions with host
M. pneumoniae can cause infections in humans, animals, plants, and cell cultures. It is a parasitic bacterium that invades the mucosal membranes of the upper and lower respiratory tract, including nasopharynx, throat, trachea, bronchi, bronchioles, and alveoli.[8] In order to survive, M. pneumoniae needs essential nutrients and compounds such as amino acids, cholesterol, precursors to nucleic acid synthesis, and fatty acids obtained from the mucosal epithelial cells of the host.[7] Its adhesion proteins attach to tracheal epithelial cells by sialoglycoproteins or sialoglycolipid receptors, which are located on its cell surface.[7] It may cause injury to the respiratory epithelial cell after its attachment. The injury of host epithelial cells caused by M. pneumoniae adhesion is thought to be due to the production of highly reactive species: hydrogen peroxide (H2O2) and superoxide radicals (O2–).[7] M. pneumoniae has the potential for intracellular localization. The intracellular existence of M. pneumoniae could facilitate the establishment of latent or chronic states, circumvent mycoplasmacidal immune mechanisms, its ability to cross mucosal barriers and gain access to internal tissues. Besides, fusion of the mycoplasmal cell membrane with that of the host not only results in the release of various hydrolytic enzymes produced by the mycoplasma, but also leads to the insertion of mycoplasmal membrane components into the host cell membrane, a process that could potentially alter receptor recognition sites and affect cytokine induction and expression.[8] As stated by Nisar et al. (2007), M. pneumonia can persist in the respiratory tract up to several months after recovery from acute pneumonia.[9] In fact, M. pneumonia can be cultured from respiratory secretions even after the pneumonia patients are treated with effective antibiotics.[9] Thus, M. pneumonia infection is chronic and persistent. Besides, Nisar et al. (2007) also adds that M. pneumonia infection causes pulmonary structural abnormalities, resulting in a decrease in expiratory flow rates and an increase in airways hyper-responsiveness in non-asthmatic individuals.[9]
Roles in asthma
M. pneumonia infection is responsible for triggering exacerbation of asthma in 3.3 to 50% in such cases.[9] Furthermore, M. pneumonia may also precede the onset of asthma, because patients with an acute infection by M. pneumonia, followed by the development of asthma, have significant improvement in lung function and asthma symptoms after they are given antimicrobial treatment against M. pneumonia. The release of proinflammatory cytokines in response to M. pneumoniae infection has been indicated as a possible mechanism leading to bronchial asthma.[8] This is because the increase of cytokine production results in a continuing inflammatory response in the airway, followed by negative effects such as immunopathological tissue damage and scarring as described in the C. pneumonia’s role in asthma section. Furthermore, patients with asthma are found to have an increased release of type II cytokines, especially IL4 and IL5, but a normal or low level of type I cytokine production. Similarly, M. pneumoniae infection promotes a T helper type 2 response, which is why M pneumoniae-positive patients with asthma have increased airways expression of tumour necrosis factor a, IL4 and IL5. The T helper type 2 predominant airways disease caused by M. pneumonia infection may lead to IgE-related hyper-responsiveness and eosinophil function, resulting in an onset of asthma.[9] There is also a possibility that M. pneumoniae infection may destroy respiratory mucosal cells and facilitate the penetration of antigens into the mucosa.[9] A study done by Laitinen et al. (1976) suggests that M. pneumoniae infection denude the epithelial surface and expose irritant receptors.[10] On top of that, M. pneumoniae induces the activation of mast cells by releasing serotonin and hexosaminidase.[8] By producing antigen, M. pneumonia is capable of initiating an antibody response. Its antigen interacts with IgE that attaches to mast cells, leading to the stimulation of histamine release followed by airway obstruction.[9]
Human rhinoviruses (HRVs)
General descriptions
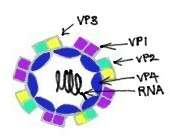
Rhinoviruses are known to be the most important common cold viruses.[11] They are ssRNA positive-strand viruses with no DNA stage, and are classified within the family Picornaviridae.[11] Rhinoviruses are small, with the length of approximately 30 nm, and do not contain an envelope.[11] Their icosahedral capsids contain 4 proteins: VP1, VP2, VP3 and VP4. VP1, VP2, and VP3 are located on the surface of the capsid and are responsible for the antigenic diversity of Rhinoviruses.[11] In contrast, VP4 is located inside the virus and its function is to anchor the RNA core to the viral capsid.[12] While sharing basic properties with enteroviruses, such as size, shape, nucleic acid composition, and ether-resistance, rhinoviruses are distinguished from enteroviruses by having a greater buoyant density and a susceptibility to inactivation if they are exposed to an acidic environment.[11] Nevertheless, they share a common ancestor with enteroviruses.[11]
Biological interactions with host
The optimal temperature for rhinovirus replication is 33-35 °C, which corresponds to the temperature of nasal mucosa. At 37 °C virus replication rate falls to 10% to 50% of optimum.[11] This may be the major reason why rhinoviruses can replicate better in the nasal passages and upper tracheobronchial tree than in the lower respiratory tract.[13] Most of the rhinovirus serotypes bind to intercellular adhesion molecule (ICAM), whereas approximately 10% of the serotypes bind to the low-density lipoprotein receptor.[13] Normally, rhinoviruses would infect small clusters of cells in the epithelial layer with little cellular cytotoxicity.[13] Although an increase in polymorphonuclear neutrophils are shown in infected nasal epithelium, little or no mucosal damage occurs from the infection.[12] Nevertheless, rhinovirus infection leads to symptoms of the common cold, which is primarily an upper airway illness.[12] Rhinovirus receptors are insensitive to neuraminidase but are sensitive to proteolytic enzymes.[11]
Roles in asthma
Asthmatic subjects in 9 to 11 years old, 80% to 85% of asthma exacerbations that were associated with reduced peak expiratory flow rates and wheezing were due to viral upper respiratory tract infections (URIs). High rates of asthma attacks due to rhinovirus infection are also found in adults.[12] It turns out that rhinovirus are capable of inducing epithelial cells to produce proinflammatory cytokines that result in airway hyperresponsiveness, neurogenic inflammatory responses, mucous secretion, inflammatory cell recruitment and activation, and plasma leakage. To support this statement, asthmatic subjects that are infected with rhinovirus have demonstrated an increase in airway hyperresponsiveness, airway obstruction, and inflammation. Similarly, rhinovirus infection has caused subjects with allergic rhinitis but no history of asthma to have a significantly increased airway hyperreactivity as well as a significantly increased incidence of late asthmatic reactions. This shows that in addition to causing airway hyperreactivity, rhinovirus also promotes the onset of non-atopic asthma.[12] Furthermore, rhinovirus infection also promotes eosinophil recruitment to airway segments after antigen challenges, and thus intensifies airway inflammatory response to antigens, leading to the development of asthma.
References
- Guilbert, T.W; Denlinger, L.C (2010). "Role of infection in the development and exacerbation of asthma". Expert Rev. Respir. Med. 4 (1): 71–83. doi:10.1586/ers.09.60. PMC 2840256. PMID 20305826.
- Hertzen, L.V. (2002). "Role of persistent infection in the control and severity of asthma: focus on Chlamydia pneumoniae". European Respiratory Journal. 19: 546–556. doi:10.1183/09031936.02.00254402.
- Coombes, Brain K (2002). "Cellular and Molecular Host-pathogen Interactions during Chlamydia Pneumoniae Infection". Open Access Dissertations and Theses. Paper 1391.
- Kou, C.C; Jackson, L.A.; Campbell, L.A.; Grayston, J.T (1995). "Chlamydia pneumoniae". Clinical Microbiology Reviews. 8 (4): 451–461.
- Larsen, R., Pogliano, K. "Chlamydophila pneumoniae". Microbe Wiki. Retrieved 24 October 2012.
- Beatty, WL; Morrison, RP.; Byrne, G. (1994). "Persistent Chlamydiae: from cell culture to a paradigm for chlamydial pathogenesis". Microbiological Reviews. 58: 686–699. PMC 372987. PMID 7854252.
- Pasternak, Y. "Mycoplasma pneumoniae". Microbe Wiki. Retrieved 24 October 2012.
- Waites, K.B.; Talkington, D. (2004). "Mycoplasma pneumoniae and Its Role as a Human Pathogen". Clinical Microbiology Reviews: 697–728.
- Nisar, N.; Guleria, R.; Kuma, S. r.; Chawla, T. C.; Biswas, N. R. (2007). "Mycoplasma pneumoniae and its role in asthma". Postgraduate Medical Journal. 83: 100–104. doi:10.1136/pgmj.2006.049023. PMC 2805928. PMID 17308212.
- Laitinen, LA; Elkin, RB.; Jacobs, L.; et al. (1976). "Changes in bronchial reactivity after administration of live attenuated influenza virus". Am. Rev. Respir. Dis.: 194–198.
- Jack Merrit Gwaltney, J. (1975). "Medical Reviews Rhinoviruses". The Yale Journal of Biology and Medicine: 17–45.
- Friedlander, S. L. (2005). "The role of rhinovirus in asthma exacerbations". J. Allergy Clin. Immunol. 116 (2): 267–273. doi:10.1016/j.jaci.2005.06.003.
- "Rhinovirus". Microbe Wiki. Retrieved 25 October 2012.