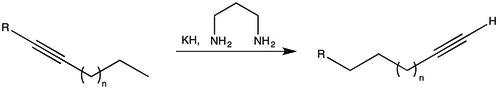
Alkyne zipper reaction
The alkyne zipper reaction is an organic reaction that involves isomerization of an internal alkyne into a terminal alkyne. This reaction was first reported by Charles Allen Brown and Ayako Yamashita in 1975.[1] The isomerization reaction proceeds for straight-chain alkynes and acetylinic alcohols. The conversion provides a useful approach for remote functionalization in long-chain alkynes.[2]
The reaction requires a strong base. The base used by Brown and Yamashita was potassium 1,3-diaminopropanide, generated in situ by adding potassium hydride to the solvent 1,3-diaminopropane.[1] Alternative approaches have been investigated due to the expensive and hazardous nature of potassium hydride; ethylenediamine has been found to be an unsuitable replacement for 1,3-diaminopropane. As an example, for the synthesis of 9-decyn-1-ol from 2-decyn-1-ol, the lithium salt of 1,3-diaminopropane in the presence of potassium tert-butoxide affords yields of approximately 85%.[2]
- HO–CH2C≡C–(CH2)6CH3 → HO(CH2)8–C≡CH
Mechanism
The alkyne zipper reaction requires a strong base, which can be generated from the reaction of potassium hydride and a diamine:[3][1][3]
The potassium 3-aminopropylamide deprotonates the less-substituted methylene adjacent to the alkyne group.[3][1]
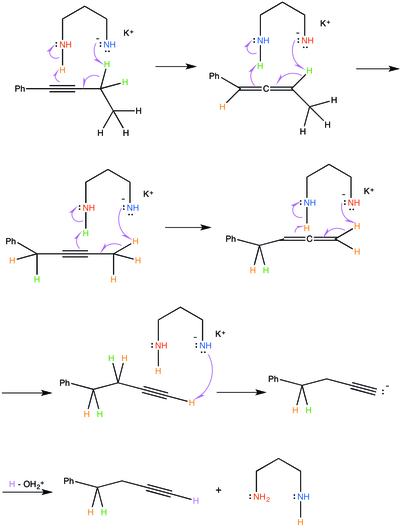
The 3-aminopropylamine anion attacks the same lesser-substituted carbon adjacent to the allene, removing a proton and catalyzing a similar process, where the electrons from the carbon-hydrogen bond move to form a triple-bond (an alkyne).[3][1] The pi-electrons that compose the neighboring double-bond in the allene are forced to attack the second amine group on the 3-aminopropylamine. In keeping with the previous step, the amine group that holds the negative charge acts as the nucleophile and the amine group that does not hold that negative charge acts as an electrophile.
These steps will be repeated, essentially moving the alkyne along the alkane chain until a terminal alkyne is achieved.[3] Once a terminal alkyne is achieved, the 3-aminopropylamine anion will attack and remove the terminal proton. However, the process stops there because the carbon-hydrogen bond electrons cannot form an additional pi-bond on top of the alkyne.[3][1] Therefore, an acetylide anion is produced. A mild acid workup will quench the acetylide anion and the 3-aminopropylamine anion.[1]
References
- C. A. Brown and A. Yamashita (1975). "Saline hydrides and superbases in organic reactions. IX. Acetylene zipper. Exceptionally facile contrathermodynamic multipositional isomeriazation of alkynes with potassium 3-aminopropylamide". J. Am. Chem. Soc. 97 (4): 891–892. doi:10.1021/ja00837a034.
- Suzanne R. Abrams and Angela C. Shaw (1988). "Triple Bond Isomerizations: 2- to 9-decyn-1-ol". Organic Syntheses. 66: 127.; Collective Volume, 8, p. 146
- "Alkyne Zipper Reaction". SynArchive. 2017. Archived from the original on 2011. Retrieved December 19, 2017.