ANNOVAR
ANNOVAR (ANNOtate VARiation) is a bioinformatics software tool for the interpretation and prioritization of single nucleotide variants (SNVs), insertions, deletions, and copy number variants (CNVs) of a given genome[1]. It has the ability to annotate human genomes hg18, hg19, hg38, and model organisms genomes such as: mouse (Mus musculus), zebrafish (Danio rerio), fruit fly (Drosophila melanogaster), roundworm (Caenorhabditis elegans), yeast (Saccharomyces cerevisiae) and many others[2]. The annotations could be used to determine the functional consequences of the mutations on the genes and organisms, infer cytogenetic bands, report functional importance scores, and/or find variants in conserved regions[2]. ANNOVAR along with SNP effect (SnpEFF) and Variant Effect Predictor (VEP) are three of the most commonly used variant annotation tools.
Background
The cost of high throughput DNA sequencing has reduced drastically from around $100 million/human genome in 2001 to around $1000/human genome in 2017.[3]. Due to this increase in accessibility, high throughput DNA sequencing has become more widely used in research and clinical settings[4][5]. Some common areas that utilize high throughput DNA sequencing extensively are: Whole Exome Sequencing, Whole Genome Sequencing (WGS), and genome wide association studies (GWAS)[6][7].
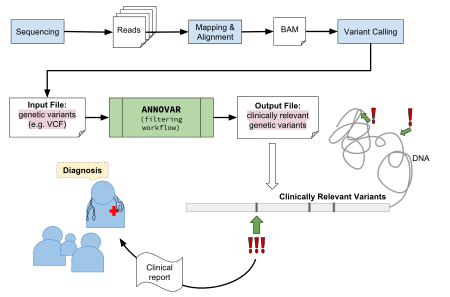
There are a growing number of tools available seeking to comprehensively manage, analyze and interpret the enormous amount of data generated from high-throughput DNA sequencing. The tools are required to be efficient and robust enough to analyze a large number of variants (more than 3 million in human genome) though sensitive enough to identify rare and clinically relevant variants that are likely harmful/deleterious[8]. ANNOVAR was developed by Dr. Kai Wang in 2010 at the Center for Applied Genomics in the University of Pennsylvania[1]. It is a type of variant annotation tool that compiles deleterious genetic variant prediction scores from programs such as PolyPhen, ClinVar, and CADD and annotates the SNVs, insertions, deletions, and CNVs of the provided genome. ANNOVAR is one of the first efficient, configurable, extensible and cross-platform compatible variant annotation tools created.
In terms of the larger bioinformatics workflow, ANNOVAR fits in near the end, after DNA sequencing reads having between mapped, aligned, and variants have been predicted from an alignment file (BAM), also known as variant calling. This process will produce a resultant VCF file, a tab-separated text file in a tabular like structure, containing genetic variants as rows. This file can then be used as input into the ANNOVAR software program for the variant annotation process, outputting interpretations of the variants identified from the upstream bioinformatics pipeline.
Types of functional annotation of genetic variants
Gene-based annotation
This approach identifies whether the input variants cause protein coding changes and the amino acids that are affected by the mutations[9]. The input file can be composed of exons, introns, intergenic regions, splice acceptor/donor sites, and 5′/3′ untranslated regions. The focus is to explore the relationship between non-synonymous mutations (SNPs, indels, or CNVs) and their functional impact on known genes[10]. Especially, gene-based annotation will highlight the exact amino acid change if the mutation is in the exonic region and the predicted effect on the function of the known gene. This approach is useful for identifying variants in known genes from Whole Exome Sequencing data.
Region-based annotation
This approach identifies deleterious variants in specific genomic regions based on the genomic elements around the gene[11]. Some categories region-based annotation will take into account are:
1) Is the variant in a known conserved genomic region?
Mutations occur during mitosis and meiosis. If there is no selective pressure for specific nucleotide sequences, then all areas of a genome would be mutated are equal rates. The genomic regions that are highly conserved indicate genomic sequences that are essential to the organism's survival and/or reproductive success. Thus, if the variant disrupts a highly conserved region, the variant is likely highly deleterious[12].
2) Is the variant in a predicted transcription factor binding site?
DNA is transcribed into messenger RNA (mRNA) by RNA polymerase II. This process can be modulated transcription factors which can enhance or inhibit binding of RNApol II. If the variant disrupts a transcription factor binding site then transcription of the gene could be altered causing changes in gene expression level and/or protein production amount. This changes could cause phenotypic variations.
3) Is the variant in a predicted miRNA target site?
MicroRNA (miRNA) is a type of RNA that complementary binds to targeted mRNA sequence to suppress or silence the translation of the mRNA. If the variant disrupts the miRNA target location, the miRNA could have altered binding affinity to the corresponding gene transcript thus changing the mRNA expression level of the transcript. This could further impact protein production levels which could cause phenotypic variations.
4) Is the variant predicted to interrupt a stable RNA secondary structure?
RNA can function at the RNA level as non-coding RNA or be translated into proteins for downstream processes. RNA secondary structures are extremely important in determining the correct half-life and function of those RNA. Two RNA species with tightly regulated secondary structures are ribosomal RNA (rRNA) and transfer RNA (tRNA) which are essential in translation of mRNA to protein. If the variant disrupts the stability of the RNA secondary structure, the half-life of the RNA could be shortened thus lowering the concentration of RNA in the cell.
Non-coding regions encompasses 99% of the human genome[13] and region-based annotation is extremely useful in identifying variants in those regions. This approach can be used on WGS data.
Filter-based annotation
This approach identifies variants that are documented in specific databases[14]. The variants could be obtained from dbSNP, 1000 Genomes Project, or user-supplied list. Additional information could be obtained from the frequency of the variants from the above databases or the predicted deleterious scores created by PolyPhen, CADD, ClinVar or many others[1]. The more infrequent a variant appears in the public database, the more deleterious it is likely to be. Results from different deleterious score prediction tools can combined together by the researcher to make a more accurate call on the variant.
Taken together, these approaches complement one another to filter through over 4 million variants in a human genome. Common, low-deleterious score variants are eliminated to reveal the rare, high-deleterious score variants which could be causal for congenital diseases.
Technical information
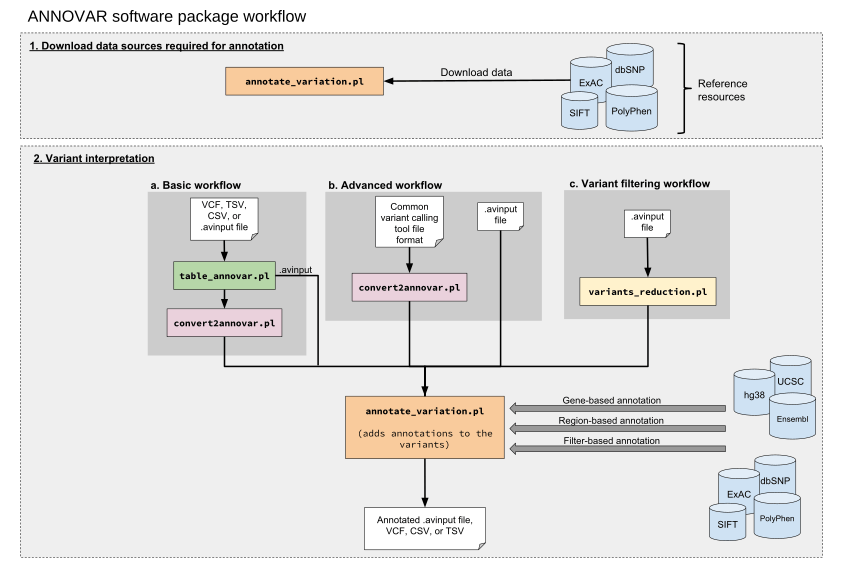
ANNOVAR is a command-line tool written in the Perl programming language and can be run on any operating system that has a Perl interpreter installed[1]. If used for non-commercial purposes, it is available free as an open-source package that is downloadable through the ANNOVAR website. ANNOVAR can process most next-generation sequencing data which has been run through a variant calling software.
| Script | Purpose | Description | Input | Output | Requirements |
|---|---|---|---|---|---|
annotate_variation.pl |
variant annotator | The core script, which functionally annotates the genetic variants via (1) gene-based, (2) region-based, and/or (3) filter-based annotation. | .avinput | .avinput | Data sources are downloaded for annotation, e.g. hg38, UCSC, 1000 Genomes Project. |
convert2annovar.pl |
file converter | Converts various file formats to the custom ANNOVAR input file format. | See "Conversion to the ANNOVAR input file format" section. | .avinput | |
table_annovar.pl |
automated variant annotator | A wrapper around annotate_variation.pl that can take VCF format along with the ANNOVAR format, performs annotation and outputs an Excel-compatible file. Ideal for beginners. |
.avinput, CSV, TSV, VCF | CSV, TSV, VCF, TXT | Data sources are downloaded for annotation, e.g. hg38, UCSC, 1000 Genomes Project. |
variants_reduction.pl |
variant reducer | Performs stepwise variant reduction on a large set of input variants to narrow down to a subset of functionally important variants. Filtering procedures include: Applies a stepwise procedure of filtering to identify subsets of variants that are likely to be related to a disease[2]. Such filtering procedures include[2]:
|
.avinput | .avinput | Gene-based annotation data sources and various filter-based annotation data sources are downloaded. |
File formats
The ANNOVAR software accepts text-based input files, including VCF (Variant Call Format), the gold standard for describing genetic loci.
The program's main annotation script, annotate_variation.pl requires a custom input file format, the ANNOVAR input format (.avinput). Common file types can be converted to ANNOVAR input format for annotation using a provided script (see below). It is a simple text file where each line in the file corresponds to a variant and within each line are tab-delimited columns representing the basic genomic coordinate fields (chromosome, start position, end position, reference nucleotides, and observed nucleotides), followed by optional columns[2]
The ANNOVAR file input contains the following basic fields:
- Chr
- Start
- End
- Ref
- Alt
For basic "out-of-the-box" usage:
A popular function of the ANNOVAR tool is the use of the table_annovar.pl script which simplifies the workflow into one single command-line call, given that the data sources for annotation have already been downloaded. File conversion from VCF file is handled within the function call, followed by annotation and output to an Excel-compatible file. The script takes a number of parameters for annotation and outputs a VCF file with the annotations as key-value pairs inside of the INFO column of the VCF file for each genetic variant, e.g. "genomic_function=exonic".
Conversion to the ANNOVAR input file format
File conversion to the ANNOVAR input format is possible using the provided file format conversion script convert2annovar.pl. The program accepts common file formats outputted by upstream variant calling tools. Subsequent functional annotation scripts annotate_variation.pl use the ANNOVAR input file. File formats that are accepted by the convert2annovar.pl include the following:[2]
- Variant Call Format
- Samtools genotype-calling pileup format
- Illumina export format from GenomeStudio
- SOLiD GFF genotype-calling format
- Complete Genomics variant format
Generating input files based on specific variants, transcripts, or genomic regions:
When investigating candidate loci that are linked to diseases, using the above variant calling file formats as input to ANNOVAR is a standard workflow for functional annotation of genetic variants outputted from an upstream bioinformatics pipeline. ANNOVAR can also be used to in other scenarios, such as interrogating a set of genetic variants of interest based on a list of dbSNP identifiers as well as variants within specific genomic or exomic regions[2].
In the case of dbSNP identifiers, providing to the convert2annovar.pl script a list of identifiers (e.g. rs41534544, rs4308095, rs12345678) in a text file along with the reference genome of interest as a parameter, ANNOVAR will output an ANNOVAR input file with the genomic coordinate fields for those variants which can then be used for functional annotation[2].
In the case of genomic regions, one can provide a genomic range of interest (e.g. chr1:2000001-2000003) along with the reference genome of interest and ANNOVAR will generate an ANNOVAR input file of all the genetic loci spanning that range. In addition, insertion and deletion size could also be specified in which the script will select all the genetic loci where a specific size of interest insertion or deletion is found.[2]
Last, if looking at variants within specific exonic regions, users can generate ANNOVAR input files for all possible variants in exons (including splicing variants) when theconvert2annovar.pl script is provided an RNA transcript identifier (e.g. NM_022162) based on the standard HGVS (Human Genome Variation Society) nomenclature.[2]
Output file
The possible output files are an annotated .avinput file, CSV, TSV, or VCF. Depending on the annotation strategy taken (see Figure below), the input and output files will differ. It is possible to configure the output file types given a specific input file, by providing the program the appropriate parameter.
For example, for the table_annovar.pl program, if the input file is VCF, then the output will also be a VCF file. If the input file is of the ANNOVAR input format type, then the output will be a TSV by default, with the option to output to CSV if the -csvout parameter is specified. By choosing CSV or TSV as the output file type, a user could open the files to view the annotations in Excel or a different spreadsheet software application. This is a popular feature among users.
The output file will contain all the data from the original input file with additional columns for the desired annotations. For example, when annotating variants with characteristics such as (1) genomic function and (2) the functional role of the coding variant, the output file will contain all the columns from the input file, followed additional columns "genomic_function" (e.g. with values "exonic" or "intronic") and "coding_variant_function" (e.g. with values "synonymous SNV" or "non-synonymous SNV").
System efficiency
Benchmarked on a modern desktop computer (3GHz Intel Xeon CPU, 8GB memory), for 4.7 million variants, ANNOVAR requires ~4 minutes to perform gene-based functional annotation, or ~15 minutes to perform stepwise "variants reduction". It is said to be practical for performing variant annotation and variant prioritization on hundreds of human genomes in a day.[2]
ANNOVAR could be sped up by using the -thread argument which enables multi-threading so that input files could be processed in parallel.
Data resources
To use ANNOVAR for functional annotation of variants, annotation datasets can be downloaded using the annotate_variation.pl script, which saves them to local disk[1]. Different annotation data sources are used for the three major types of annotation (gene-based, region-based, and filter-based).
These are some of the data sources for each annotation type:
Region-based annotation[11]
- ENCODE
- Custom-made databases conforming to GFF3 (Generic Feature Format version 3)
Filter-based annotation[14]
| 1000 Genomes Project | LRT | ClinVar |
| dbSNP | MutationTaster | CADD |
| avSNP | GERP++ | DANN |
| dbNSFP | ExAC | COSMIC |
| SIFT | ESP (Exome Sequencing Project) | ICGC |
| PolyPhen 2 | gnomAD allele frequency | NCI60 |
| PhyloP | Complete Genomics allele frequency |
Given the large number of data sources for filter-based annotation, here are examples of which subsets of the datasets to use for a few of the most common use cases.[14]
- For frequency of variants in whole-exome data:[14]
- ExAC: with allele frequencies for all ethnic groups
- NHLBI-ESP: from 6500 exomes, use three population groupings
- gnomAD allele frequency: with allele frequencies for multiple populations
- For disease-specific variants:[14]
- ClinVar: with individual columns for each ClinVar field for each variant
- COSMIC: somatic mutations from cancer and the frequency of occurrence in each subtype of cancer
- ICGC: mutations from the International Cancer Genome Consortium
- NCI-60: human tumor cell panel exome sequencing allele frequency data
Example application
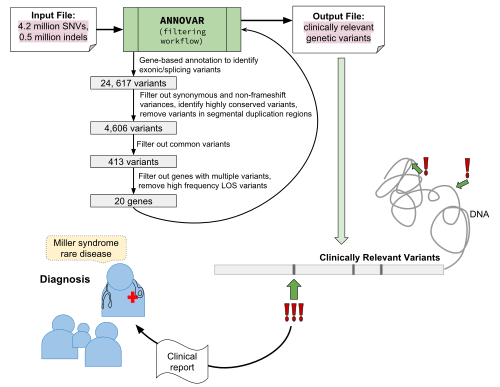
Using ANNOVAR for prioritization of genetic variants to identify mutations in a rare genetic disease
ANNOVAR is one of the common annotation tools for identifying candidate and causal mutations and genes for rare genetic diseases.
Using a combination of gene-based and filter-based annotation followed by variant reduction based on the annotation values of the variants, the causal gene in a rare recessive Mendelian disease called Miller syndrome can be identified.[1]
This will involve synthesizing a genome-wide data set of ~4.2 million single nucleotide variants (SNVs) and ~0.5 million insertions and deletions (indels).[1]. Two known causal mutations for Miller syndrome (G152R and G202A in the DHODH gene) are also included[1]
Steps in identifying the causal variants for the disease using ANNOVAR:[1]
- Gene-based annotation to identify exonic/splicing variants of the combination of SNVs and indels (~4.7 million variants) where a total of 24,617 exonic variants are identified.[1]
- Since Miller syndrome is a rare Mendelian disease, exonic protein-changing variants are of interest only, which makes up 11,166.[1]. From that, 4860 variants are identified that falls in highly conserved genomic regions[1]
- As public databases such as dbSNP and 1000 Genomes Project archive previously reported variants which are often common, it is less likely that they will contain the Miller syndrome causal variants which are rare[1]. Hence, variants found in those data sources are filtered out and 413 variants remain.
- Then, genes are assessed for whether multiple variants exist in the same gene as compound heterozygotes and 23 genes are left.[1]
- Finally, ‘dispensable’ genes are removed, those which have high-frequency non-sense mutations (in greater than 1% of subjects in the 1000 Genomes Project) which are susceptible to sequencing and alignment errors in short-read sequencing platform.[1]. These genes are considered less likely to be causal of a rare Mendelian disease. Three genes as result are filtered out, and 20 candidate genes are leftover, including the causal gene DHODH[1]
Limitations of ANNOVAR
Two limitations of ANNOVAR relate to detection of common diseases and larger structural variant annotations. These problems are present in all current variant annotation tools.
Most common diseases such as diabetes and Alzheimer have multiple variants throughout the genome which are common in the population[15][16]. These variants are expected to have low individual deleterious scores and cause disease though the accumulation of multiple variants. However ANNOVAR has default "variant-reduction" schemes that provides a small list of rare and highly predicted deleterious variants[17]. These default settings could be optimized so the output data would display additional variants with decreasing predicted deleterious scores[2]. ANNOVAR is primarily used for identifying variants involved rare diseases where the causal mutation is expected to be rare and highly deleterious.
Larger structural variants (SVs) such as chromosomal inversions, translocations, and complex SVs have been shown to cause diseases such as haemophilia A and Alzheimer's.[18][19]. However, SVs are often difficult to annotate because it is difficult to assign specific deleterious scores to large mutated genomic regions. Currently, ANNOVAR can only annotate genes contained within deletions or duplications, or small indels of <50bp. ANNOVAR cannot infer complex SVs and translocations[17]
Alternate variant annotation tools
There are also two other types of SNP annotation tools that are similar to ANNOVAR: SNP effect (SnpEFF) and Variant Effect Predictor (VEP). Many of the features between ANNOVAR, SnpEFF, and VEP are the same including the input and output file format, regulatory region annotations, and know variant annotations. However, the main differences are that ANNOVAR cannot annotate for loss of function predictions whereas both SnpEFF and VEP can. Also, ANNOVAR cannot annotate microRNA structural binding locations whereas VEP can[20]. MicroRNA structural binding location predictions can be informative in revealing post-transcriptional mutations’ role in disease pathogenesis.[21] Loss of function mutations are changes in the genome that results in the total dysfunction of the gene product. Thus, these predictions could be extremely informative in regards to disease diagnosis, especially in rare monogenic diseases.
| Class | Feature | VEP | Annovar | SnpEff |
| General | Availability | Free | Free (academic use only) | Free |
| Input | VCF | Yes | Yes | Yes |
| Sequence variants | Yes | Yes | Yes | |
| Structural variants | Yes | Yes | Yes | |
| Output | VCF | Yes | Yes | Yes |
| Transcript sets | Ensembl | Yes | Yes | Yes |
| RefSeq | Yes | Yes | Yes | |
| User-created databases | Yes | Yes | Yes | |
| Interfaces | Local package | Yes | Yes | Yes |
| Instant prediction web interface | Yes | No | No | |
| Consequence types | Splicing predictions | Yes (via plugins) | Yes (via external data) | Yes (experimental) |
| Loss of function prediction | Yes (via plugins) | No | Yes | |
| Non-coding | Regulatory features | Yes | Yes | Yes |
| Support multiple cell lines | Yes | No | Yes | |
| miRNA structure location | Yes (via plugins) | No | No | |
| Known variants | Report known variants | Yes | Yes | Yes |
| Filter by frequency | Yes | Yes | Yes | |
| Clinical significance | Yes | Yes | Yes | |
| Other filters | Pre-set filters | Yes | Yes | Yes |
*Table adapted from McLaren et al. (2016).
References
- Hakonarson, Hakon; Li, Mingyao; Wang, Kai (2010-09-01). "ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data". Nucleic Acids Research. 38 (16): e164. doi:10.1093/nar/gkq603. ISSN 0305-1048. PMC 2938201. PMID 20601685.
- "ANNOVAR website". www.openbioinformatics.org. Retrieved 2019-02-28.
- "DNA Sequencing Costs: Data". National Human Genome Research Institute (NHGRI). Retrieved 2019-04-04.
- Emerson, Ryan O.; Sherwood, Anna M.; Rieder, Mark J.; Guenthoer, Jamie; Williamson, David W.; Carlson, Christopher S.; Drescher, Charles W.; Tewari, Muneesh; Bielas, Jason H. (December 2013). "High-throughput sequencing of T cell receptors reveals a homogeneous repertoire of tumor-infiltrating lymphocytes in ovarian cancer". The Journal of Pathology. 231 (4): 433–440. doi:10.1002/path.4260. ISSN 0022-3417. PMC 5012191. PMID 24027095.
- Blayney, Jaine K.; Parkes, Eileen; Zheng, Huiru; Taggart, Laura; Browne, Fiona; Haberland, Valeriia; Lightbody, Gaye (2018). "Review of applications of high-throughput sequencing in personalized medicine: barriers and facilitators of future progress in research and clinical application". Briefings in Bioinformatics. doi:10.1093/bib/bby051. PMID 30084865.
- Reference, Genetics Home. "What are whole exome sequencing and whole genome sequencing?". Genetics Home Reference. Retrieved 2019-04-04.
- Reference, Genetics Home. "What are genome-wide association studies?". Genetics Home Reference. Retrieved 2019-04-04.
- The 1000 Genomes Project Consortium (October 2015). "A global reference for human genetic variation". Nature. 526 (7571): 68–74. Bibcode:2015Natur.526...68T. doi:10.1038/nature15393. ISSN 1476-4687. PMC 4750478. PMID 26432245.
- "Gene-based Annotation - ANNOVAR Documentation". annovar.openbioinformatics.org. Retrieved 2019-02-28.
- Yang, Hui; Wang, Kai (October 2015). "Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR". Nature Protocols. 10 (10): 1556–1566. doi:10.1038/nprot.2015.105. ISSN 1754-2189. PMC 4718734. PMID 26379229.
- "Region-based Annotation - ANNOVAR Documentation". annovar.openbioinformatics.org. Retrieved 2019-02-28.
- Jordan, I. King; Rogozin, Igor B.; Wolf, Yuri I.; Koonin, Eugene V. (June 2002). "Essential Genes Are More Evolutionarily Conserved Than Are Nonessential Genes in Bacteria". Genome Research. 12 (6): 962–968. doi:10.1101/gr.87702. ISSN 1088-9051. PMC 1383730. PMID 12045149.
- Reference, Genetics Home. "What is noncoding DNA?". Genetics Home Reference. Retrieved 2019-03-01.
- "Filter-based Annotation - ANNOVAR Documentation". annovar.openbioinformatics.org. Retrieved 2019-02-28.
- Wu, Yiming; Jing, Runyu; Dong, Yongcheng; Kuang, Qifan; Li, Yan; Huang, Ziyan; Gan, Wei; Xue, Yue; Li, Yizhou (2017-03-06). "Functional annotation of sixty-five type-2 diabetes risk SNPs and its application in risk prediction". Scientific Reports. 7: 43709. Bibcode:2017NatSR...743709W. doi:10.1038/srep43709. ISSN 2045-2322. PMC 5337961. PMID 28262806.
- Emahazion, T.; Feuk, L.; Jobs, M.; Sawyer, S. L.; Fredman, D.; St Clair, D.; Prince, J. A.; Brookes, A. J. (July 2001). "SNP association studies in Alzheimer's disease highlight problems for complex disease analysis". Trends in Genetics. 17 (7): 407–413. doi:10.1016/S0168-9525(01)02342-3. ISSN 0168-9525. PMID 11418222.
- Yang, Hui; Wang, Kai (October 2015). "Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR". Nature Protocols. 10 (10): 1556–1566. doi:10.1038/nprot.2015.105. ISSN 1754-2189. PMC 4718734. PMID 26379229.
- Lakich, Delia; Kazazian, Haig H.; Antonarakis, Stylianos E.; Gitschier, Jane (November 1993). "Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A". Nature Genetics. 5 (3): 236–241. doi:10.1038/ng1193-236. ISSN 1061-4036. PMID 8275087.
- Lupski, James R. (June 2015). "Structural Variation Mutagenesis of the Human Genome: Impact on Disease and Evolution". Environmental and Molecular Mutagenesis. 56 (5): 419–436. doi:10.1002/em.21943. ISSN 0893-6692. PMC 4609214. PMID 25892534.
- McLaren, William; Gil, Laurent; Hunt, Sarah E.; Riat, Harpreet Singh; Ritchie, Graham R. S.; Thormann, Anja; Flicek, Paul; Cunningham, Fiona (2016-06-06). "The Ensembl Variant Effect Predictor". Genome Biology. 17 (1): 122. doi:10.1186/s13059-016-0974-4. ISSN 1474-760X. PMC 4893825. PMID 27268795.
- Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y (January 2009). "miR2Disease: a manually curated database for microRNA deregulation in human disease". Nucleic Acids Research. 37. 37 (Database issue): D98–104. doi:10.1093/nar/gkn714. PMC 2686559. PMID 18927107.